Scientific publications produced through the center’s research activities and collaborations.
To jump to previous years or all publications from a C3 PI, please use the buttons below to navigate the page.
Thaddeus W. Golbek, Mette H. Rasmussen, Mikkel Bregnhøj, Thomas Boesen, Taner Drace, Ryan Faase, Joe E. Baio, and Tobias Weidner
Specialized bacteria can effectively nucleate ice crystals using ice nucleating proteins (INPs) anchored to the cell surface. Biogenic freezing has several applications, from snow making to cryo-medicine and freeze/antifreeze materials. For biomimetic designs of INP analogs, it is important to understand how the proteins involved in the process bind to material surfaces. In this study, we determine the binding of a model INP to hydrophobic self-assembled monolayers (SAMs) as well as hydrophilic carboxyl-terminated SAMs. INPs are large proteins with more than 1200 amino acids and a long series of repeat units. Since full-length INPs are difficult to produce and handle, we have investigated a shorter model INP dubbed InaZ9R, which has nine repeat units and still folds into the hallmark beta-helix structure known from the full-length protein. Combining x-ray photoelectron spectroscopy and near-edge x-ray absorption fine structure spectroscopy, we find that InaZ9R form closely packed monolayers on both hydrophilic and hydrophobic surfaces. Angle-resolved nitrogen K-edge near-edge x-ray absorption fine structure spectra show a high degree of orientational order associated with the native β-sheet structure.
Full citation: Thaddeus W. Golbek, Mette H. Rasmussen, Mikkel Bregnhøj, Thomas Boesen, Taner Drace, Ryan Faase, Joe E. Baio, Tobias Weidner, Binding and orientation of ice nucleating proteins on hydrophilic and hydrophobic surfaces probed by photoelectron spectroscopies, Biointerphases, 21, 3, 031002 (2026). https://doi.org/10.1116/6.0005223
Ivo Neefjes, Jakub Kubečka, and Jonas Elm
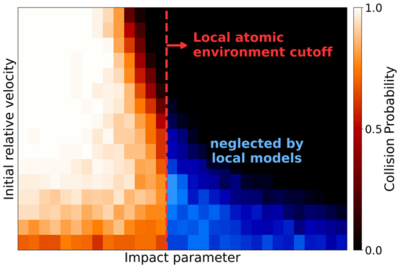
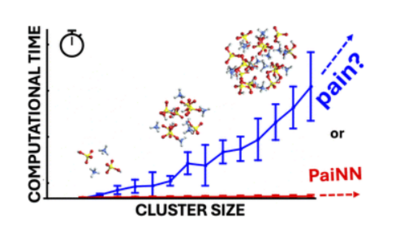
Molecular collisions and subsequent clustering events are fundamental to atmospheric cluster formation. Accurately modeling these processes requires interatomic potentials that simultaneously capture the long-range forces governing collision kinetics and the short-range quantum effects driving reactivity. In this work, we evaluate the AIMNet2 and PaiNN machine learning architectures trained on GFN1-xTB and ωB97X-3c quantum chemical data for molecular collisions involving sulfuric acid.
The models exhibit low mean absolute errors in energies and forces and accurately reproduce potentials of mean force relative to the GFN1-xTB reference. However, discrepancies are observed for the collision dynamics. While AIMNet2 accurately reproduces reference collision rate coefficients across all systems, PaiNN underestimates the rate coefficient for the charged sulfuric acid–bisulfate system by ∼ 50 %. This error originates from the model's local atomic environment approximation, which neglects the strong long-range attractive forces at large intermolecular distances. Simulations with the OPLS-AA classical force field demonstrate that simple fixed partial charges are sufficient to describe these interactions.
Comparing models trained on GFN1-xTB and ωB97X-3c data reveals that while increasing the level of electronic structure theory significantly alters the potential energy surface in the short-range binding region, it generally has less impact on the long-range shoulder and the resulting collision rate coefficients.
Our results highlight that while local equivariant models like PaiNN offer exceptional accuracy for thermodynamics, correctly simulating collision kinetics in systems with strong long-range interactions requires models that explicitly account for forces beyond the local environment, such as AIMNet2.
Full citation: Ivo Neefjes, Jakub Kubečka, and Jonas Elm, Machine learning interatomic potentials with accurate long-range interactions for molecular dynamics collision simulations of atmospherically-relevant molecules, Atmos. Chem. Phys., 26, 7631–7645 (2026). https://doi.org/10.5194/acp-26-7631-2026
Galib Hasan, Theo Kurtén, Ivo Neefjes, and Jonas Elm
Oligomerization reactions from RO2 + R′O2 radicals, occurring via a triplet (RO···3O2···OR′) cluster, are an important gas-phase reaction for the formation of low-volatile ROOR′ accretion products. However, it remains unknown whether such reactions can occur at the interface of freshly nucleated particles (FNPs). For instance, FNPs coated with a shell of organic compounds could potentially form accretion products at the surface, further stabilizing the particle. Using quantum chemical methods, we here study how the RO2 + R′O2 reaction is influenced by interaction with FNP precursors such as sulfuric acid (SA), ammonia (AM), and dimethylamine (DMA). For the RO2′s, we tested simple branched hydroxyl peroxy radicals (HO–RO2) as the tether to the FNP components. Cluster structures were obtained using a systematic conformational sampling approach based on the ABCluster program and CREST. We calculated the final structure and vibrational frequencies at the ωB97X-D/6–31++G(d,p) level of theory. Energy levels for intersystem crossing calculations were carried out at the XMC-QDPT2/6–311++G(d,p) level of theory, and spin–orbit coupling matrix elements were calculated using CASSCF(6,4)/6–311++G(d,p). Our calculations show that the rate of the intersystem crossing needed to form ROOR′ accretion products at the FNP model clusters lies in the range of 106–109 s–1, similar to the rate previously computed in the gas phase. We also find that both the intermediate (RO···3O2···OR′) clusters and the resulting ROOR′ accretion products interact strongly with the FNP components, leading to suppressed evaporation if formed at the surface. Unfortunately, the formation is limited by the requirement of two RO2 radicals being involved. We hypothesize a pathway where the RO2/R′O2 are formed via oxidation reactions at the surface and recombine via a Langmuir–Hinshelwood mechanism. However, this must still be considered an extremely rare event.
Full citation: Galib Hasan, Theo Kurtén, Ivo Neefjes, and Jonas Elm, Can Accretion Products Be Formed at the Interface of Freshly Nucleated Particles?, ACS Omega, 11, 14, 22099-22109 (2026). https://doi.org/10.1021/acsomega.5c13316
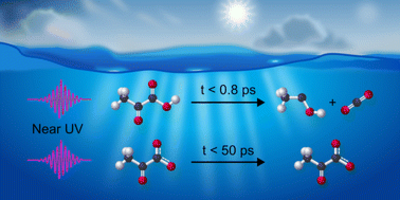
Jan Thøgersen, Tobias Weidner, Frank Jensen
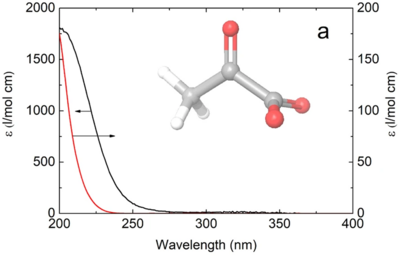
Near-UV photolysis of pyruvic acid results in decarboxylation within 0.8 ps. In contrast, pyruvate anions excited by near-UV light relax back to the ground state within 50 ps with no detectable photolysis. The sharply different behavior of the acid and its conjugate base provides a mechanistic explanation for the puzzling pH dependence of the pyruvic acid photolysis.
Full citation: Jan Thøgersen, Tobias Weidner, Frank Jensen, The primary near-UV photochemistry of aqueous pyruvic acid, Phys. Chem. Chem. Phys., 28, 7830-7834 (2026). https://doi.org/10.1039/d5cp04995d
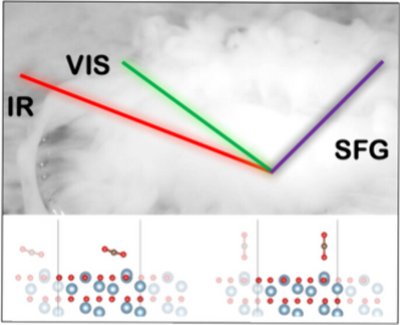
Mikkel Bregnhøj, Kris Strunge, Ivo Neefjes, Jonas Elm, and Tobias Weidner
The interaction between liquid carbon dioxide and material surfaces plays an important role for the function and durability of carbon capture and utilization (CCU) systems. Experimental data on this interaction are scarce due to the difficulty in tracking interfacial layers of CO2 in contact with solid surfaces. In this work, we use surface-sensitive sum-frequency generation (SFG) spectroscopy combined with molecular dynamics simulations and DFT calculations to measure the geometry and orientation of the first molecular layer of liquid CO2 in contact with a sapphire surface. The results show that the CO2 molecules preferentially adopt a tilted orientation with an angle of 14 ± 8° with respect to the surface normal. Furthermore, the electronic structure of CO2 is perturbed by the interface leading to symmetry breaking: one C═O bond is elongated along the molecular axis while the other is shortened. These results pave the way for using SFG spectroscopy to measure the interactions of liquid CO2 with functional surfaces, catalysts, and CCU relevant materials.
Full citation: Mikkel Bregnhøj, Kris Strunge, Ivo Neefjes, Jonas Elm, and Tobias Weidner, Surface Spectroscopy Reveals Ordering and Alignment of Liquid Carbon Dioxide at Sapphire Interfaces, The Journal of Physical Chemistry Letters, 17 (10), 2969-2973 (2026). https://doi.org/10.1021/acs.jpclett.5c03771
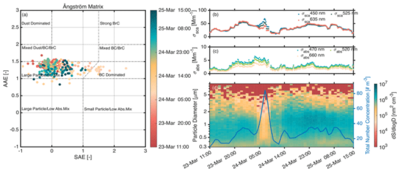
Ulas Im, Bjørn H. Samset, Athanasios Nenes, Jennie L. Thomas, Harri Kokkola, Oleg Dubovik, Vassilis Amiridis, Antti Arola, Nicolas Bellouin, Angela Benedetti, Merete Bilde, Sara Blichner, Stefano Decesari, Annica M. L. Ekman, Carlos Pérez García-Pando, Silke Gross, Edward Gryspeerdt, Otto Hasekamp, Ralph A. Kahn, Anton Laakso, Ulrike Lohmann, Louis Marelle, Andreas H. Massling, Cathrine Lund Myhre, Mira Pöhlker, Johannes Quaas, Tomi Raatikainen, Ilona Riipinen, Julia Schmale, Patric Seifert, Henrik Skov, Chris Smith, Moa K. Sporre, Philip Stier, Trude Storelvmo, Kostas Tsigaridis, Bastiaan van Diedenhoven, Annele Virtanen, Ulla Wandinger, Laura J. Wilcox, and Paul Zieger
Aerosol-cloud interactions (ACI) are a major source of uncertainty in climate science, critically affecting our ability to project near-term climate evolution and assess societal risks. These interactions influence effective radiative forcing, cloud dynamics, and precipitation patterns, yet remain insufficiently constrained due to limitations in observations, modeling, and process understanding. This uncertainty hampers robust policy advice across multiple domains—from estimating remaining carbon budgets and climate sensitivity, to anticipating regional extreme events and evaluating climate interventions such as solar radiation modification. In many cases, the influence of ACI is either underappreciated or excluded from decision-making frameworks due to its complexity and lack of quantification. This perspective outlines a path forward to overcome these barriers by leveraging emerging opportunities in satellite remote sensing, ground-based and airborne observations, high-resolution climate modeling, and machine learning. We identify key areas where rapid progress is feasible, including improved retrievals of cloud microphysical properties, better representation of natural aerosols in a warming world, and enhanced integration of observational and modeling communities. Even as anthropogenic aerosol and its impacts on clouds is reducing owing to emissions controls, addressing ACI uncertainties remains essential for refining climate projections, supporting effective mitigation and adaptation strategies, and delivering actionable science to policymakers in a rapidly changing climate system.
Full citation: Ulas Im, Bjørn H. Samset, Athanasios Nenes, Jennie L. Thomas, Harri Kokkola, Oleg Dubovik, Vassilis Amiridis, Antti Arola, Nicolas Bellouin, Angela Benedetti, Merete Bilde, Sara Blichner, Stefano Decesari, Annica M. L. Ekman, Carlos Pérez García-Pando, Silke Gross, Edward Gryspeerdt, Otto Hasekamp, Ralph A. Kahn, Anton Laakso, Ulrike Lohmann, Louis Marelle, Andreas H. Massling, Cathrine Lund Myhre, Mira Pöhlker, Johannes Quaas, Tomi Raatikainen, Ilona Riipinen, Julia Schmale, Patric Seifert, Henrik Skov, Chris Smith, Moa K. Sporre, Philip Stier, Trude Storelvmo, Kostas Tsigaridis, Bastiaan van Diedenhoven, Annele Virtanen, Ulla Wandinger, Laura J. Wilcox, and Paul Zieger, Aerosol-cloud interactions: Overcoming a barrier to projecting near-term climate evolution and risk. AGU Advances, 7, e2025AV001872 (2026). https://doi.org/10.1029/2025AV001872
Zihui Teng, Jane Tygesen Skønager, Andreas Massling, Henrik Skov, Nikolaos Evangeliou, Sabine Eckhardt, Merete Bilde, and Bernadette Rosati
Coastal aerosols are formed through the complex mixing between marine air masses and continental emissions, which originate from both natural and anthropogenic sources. The properties of coastal aerosols are decisive for their interaction with sunlight and their influence on clouds, as well as the potential health implications for the population in these areas. In this study, the aerosol properties and sources at Aarhus Bay, Denmark, were investigated by combining in situ aerosol light scattering and absorption with size distribution measurements and footprint analysis by FLEXPART. Our analysis demonstrates a considerable contribution of anthropogenic aerosols from both fossil fuel combustion and biomass burning, as well as periods with highly scattering aerosols. Furthermore, good agreement was found between in situ and modelled black-carbon data. Combining in situ measurements and FLEXPART analysis further evidenced a major impact of local emissions, as well as a few long-range transport intrusions.
Full citation: Zihui Teng, Jane Tygesen Skønager, Andreas Massling, Henrik Skov, Nikolaos Evangeliou, Sabine Eckhardt, Merete Bilde, and Bernadette Rosati, Characterizing aerosol sources based on aerosol optical properties and dispersion modelling in a Scandinavian Coastal Area (Aarhus, Denmark), Aerosol Research, 4, 169–187 (2026). https://doi.org/10.5194/ar-4-169-2026
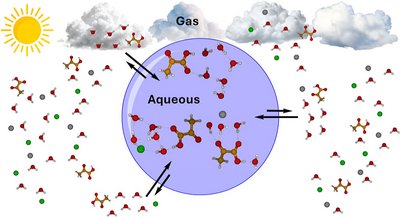
Georg Baadsgaard Trolle, Jakub Kubečka, and Jonas Elm
Organic acids are important atmospheric compounds that affect the aerosol physicochemical properties and the formation of secondary organic aerosols (SOA) with implications for air quality and climate. Pyruvic acid (PA) is ubiquitous in the atmosphere, biosphere, and hydrosphere. While the pure gas-phase and aqueous-phase chemistry of PA has been extensively studied, its simultaneous interactions with water and ions in the particle phase remains elusive. Here, we present a study on the solvation of PA and its structurally similar analogs─lactic acid (LA), propionic acid (ProA), and 2,2-dihydroxypropionic acid (diol)─by probing geometries, solvation free energies, and infrared (IR) absorption spectra using quantum chemical methods. We performed a refinement of structures in the aqueous phase based on an elaborate configurational sampling scheme in the gas phase, which we have reported previously. The aqueous phase is modeled using explicit microhydration within an implicit polarizable continuum model. We find that the solvated organic acid clusters have a high conservation of geometry when transitioning from the gas phase to the aqueous particle phase, while the solvated ion-containing clusters show significantly larger structural rearrangements. Solvation of organic acids is found to be thermodynamically favorable in the aqueous particle phase─both with and without ions─unlike in the gas phase. Finally, in order to identify weakly bound clusters and guide future experiments, our IR absorption analysis shows that the harmonic frequencies of PA carboxylic O–H stretching of the microhydrated PA clusters are red-shifted in the spectrum in the aqueous phase compared to the gas phase. Conversely, we find no clear trends in the spectrum obtained with our qualitative approach for the O–H frequencies of the microhydrated ion-containing PA clusters.
Full citation: Georg Baadsgaard Trolle, Jakub Kubečka, and Jonas Elm, Modeling the Aerosol Aqueous Phase: Solvation of Pyruvic Acid Analogs and Na+, Cl– Ions, The Journal of Physical Chemistry A, 130 (5), 1175-1187 (2026). https://doi.org/10.1021/acs.jpca.5c07598
Bernadette Rosati, Jane Tygesen Skønager, Marat Bektassov, Zihui Teng, Marianne Glasius, Marta Barbato, Merete Bilde, Kasper Vita Kristensen, and Sylvie V. M. Tesson
Microalgae emit volatile organic compounds (VOCs) that can profoundly impact climate by leading to new particle formation and influencing clouds. Among these VOCs, dimethyl-sulphide (DMS) is of particular interest due to its key role in atmospheric processes. Despite its importance, many detailed processes linking microalgae and sea-atmosphere interactions remain poorly understood. We investigated the response of a freshwater and saltwater microalgal species of haptophytes known to produce DMS, to air entrainment and bubble-bursting mechanisms relevant for wave-breaking over the ocean. We show that bubbling resulted in the successful aerosolisation of microalgae and concurrent emission of DMS. In contrast, only background levels of DMS were detected when bubbling ceased, suggesting a critical role of bubbles in the sea-air exchange of DMS under the studied conditions. DMS mixing ratios were not correlated with the emitted particle concentrations and decreased over time, while particle concentrations remained stable. Bubbling also significantly reduced the viability of aquatic microalgae. Approximately half of the aerosolised microalgae were viable upon emission, but were not able to grow during subsequent cultivation recovery. Thus, the potential for microalgae to disperse to new environments via aerosolization is low, while their climate impact through the release of DMS remains substantial.
Full citation: Bernadette Rosati, Jane Tygesen Skønager, Marat Bektassov, Zihui Teng, Marianne Glasius, Marta Barbato, Merete Bilde, Kasper Vita Kristensen, and Sylvie V. M. Tesson, Aerosolisation of microalgae: unveiling dimethyl-sulfide emissions during bubbling, npj Clim Atmos Sci 9, 32 (2026). https://doi.org/10.1038/s41612-025-01305-4
Daniel Ayoubi, Galib Hasan, Luís P. Viegas, Jakub Kubečka, and Jonas Elm
The gas-phase hydrogen abstraction reaction kinetics of atmospheric volatile organic compounds (VOCs) have been investigated using multiconformer transition state theory (MC-TST) as part of the development of the Jammy Key for Transition States (JKTS), an automated tool developed to address the vast number of organic species in the atmosphere that constantly undergo reactions with radicals. The rate constants for OH-initiated reactions with several short-chain compounds─methane, ethane, propane, and their corresponding alcohols and carbonyls─were computationally determined and compared to experimental data. Additionally, the OH abstraction kinetics of pinonaldehyde, a key oxidation product of biogenic VOCs, were studied in detail. Tunnelling effects were evaluated using Wigner and Eckart tunnelling corrections to ensure accurate prediction of reaction rates. JKTS yielded rate constants within a factor of ∼2–3 of experimental data across all systems studied, with branching ratios for pinonaldehyde showing significant contributions from aldehydic and tertiary hydrogen abstraction pathways. The calculated rate constants for pinonaldehyde, 1.739 × 10–11 cm3 molecule–1 s–1 (Eckart) and 1.847 × 10–11 cm3 molecule–1 s–1 (Wigner), align well with the experimental values of (4–9) × 10–11 cm3 molecule–1 s–1 at room temperature. These results demonstrate the capability of JKTS to automate the computation of reaction kinetics and support its application in atmospheric chemistry for accurate modeling of VOC oxidation mechanisms.
Full citation: Daniel Ayoubi, Galib Hasan, Luís P. Viegas, Jakub Kubečka, and Jonas Elm, Automatization of Atmospheric OH Radical Abstraction Reactions, The Journal of Physical Chemistry A, 130 (4), 914-926 (2026). https://doi.org/10.1021/acs.jpca.5c07873
Ivo Neefjes, Yosef Knattrup, Haide Wu, Georg Baadsgaard Trolle, Jonas Elm, and Jakub Kubečka
To improve computational modeling of hydrated atmospheric molecular clusters, we systematically evaluated quantum-chemical methods for predicting accurate structural and energetic properties of clusters containing a variety of atmospherically relevant acids and bases, with up to five water molecules. We find that the commonly applied ωB97X-D/6-31++G(d,p) method with DLPNONormalPNO–CCSD(T0)/aug-cc-pVTZ electronic energy correction is suitable for hydrated clusters. Composite density functional methods such as B97-3c, r2SCAN-3c, and ωB97X-3c are effective for pre-screening or modeling large clusters, while the local natural orbital approach LNO–CCSD(T)/aug′-cc-pVTZ is well suited for accurate refinement due to its low memory requirements, high accuracy, and favorable computational scaling. Nevertheless, the ωB97X-3c method has a reasonable accuracy even without the electronic energy correction.
We also assessed thermochemical corrections beyond the conventional harmonic oscillator approximation applied only to the lowest free-energy structure. For the limiting cases of no corrections and the ideal maximum corrections, we calculated hydration distributions and particle formation rates, with a specific emphasis on sulfuric acid–ammonia (SA–AM), sulfuric acid–dimethylamine (SA–DMA), and methanesulfonic acid–methylamine (MSA–MA) clusters. Hydration of small clusters is generally limited, with only selected SA- and MSA-containing clusters showing substantial hydration. Due to the high water concentration in the atmosphere, hydration equilibrates quickly, increasing the number of accessible states and thus stabilizing clusters. However, its effect on cluster formation and new particle formation is highly system-dependent.
MSA–MA particle formation rates are more sensitive to hydration than those of SA–AM or SA–DMA, though the enhancement remains modest. Despite being more hydrated than SA–DMA clusters, MSA–MA clusters form new particles at relatively low rates, comparable to SA–AM. Under typical atmospheric conditions, SA–DMA is expected to dominate new particle formation, even at high humidity.
Full citation: Neefjes, I., Knattrup, Y., Wu, H., Trolle, G. B., Elm, J., and Kubečka, J., Thermodynamic benchmarking of hydrated atmospheric clusters in early particle formation, Aerosol Research, 4, 1–22, (2026). https://doi.org/10.5194/ar-4-1-2026
Lauri Seppäläinen, Jakub Kubečka, Jonas Elm, and Kai R. Puolamäki
Understanding how atmospheric molecular clusters form and grow is key to resolving one of the biggest uncertainties in climate modeling: the formation of new aerosol particles. While quantum chemistry offers accurate insights into these early-stage clusters, its steep computational costs limit large-scale exploration. In this work, we present a fast, interpretable, and surprisingly powerful alternative: the k-nearest neighbor (k-NN) regression model. By leveraging chemically informed distance metrics, including a kernel-induced metric and one learned via metric learning for kernel regression (MLKR), we show that simple k-NN models can rival more complex kernel ridge regression (KRR) models in accuracy while reducing computational time by orders of magnitude. We perform this comparison with the well-established Faber–Christensen–Huang–Lilienfeld (FCHL19) molecular descriptor; however, other descriptors (e.g., FCHL18, MBDF, and CM) can be shown to have similar performance. Applied to both simple organic molecules in the QM9 benchmark set and large data sets of atmospheric molecular clusters (sulfuric acid–water and sulfuric–multibase–base systems), our k-NN models achieve near-chemical accuracy, scale seamlessly to data sets with over 250,000 entries, and even appears to extrapolate to larger unseen clusters with minimal error (often nearing 1 kcal/mol). With built-in interpretability and straightforward uncertainty estimation, this work positions k-NN as a potent tool for accelerating discovery in atmospheric chemistry and beyond.
Full citation: Lauri Seppäläinen, Jakub Kubečka, Jonas Elm, and Kai R. Puolamäki, Fast and Interpretable Machine Learning Modeling of Atmospheric Molecular Clusters, The Journal of Physical Chemistry A, 130 (4), 902-913, (2026). https://doi.org/10.1021/acs.jpca.5c06950
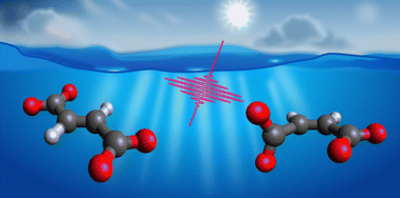
Jan Thøgersen, Mikkel Bregnhøj, Tobias Weidner and Frank Jensen
Molecular symmetry can influence the photochemical fate of molecules by controlling excited-state lifetimes and, consequently, the time window available for secondary reactions. We have investigated the primary photodynamics of aqueous fumarate and maleate, the trans- and cis-isomers of 2-butenedioate, using femtosecond transient infrared absorption spectroscopy. Following π* ← π excitation at 200 nm, both isomers undergo rapid decarboxylation to form CO2 and acrylate with identical quantum yields of maximum Φ = 30 ± 10%, independent of symmetry. Strikingly, excited-state lifetimes differ by more than an order of magnitude: fumarate remains excited for 5.9 ps, while maleate returns to its ground state in less than 0.5 ps. We attribute this disparity to the higher symmetry of fumarate, which may restrict nonradiative decay pathways. These results demonstrate that even when primary photoproducts are unaffected by symmetry, excited-state lifetimes – and thus the potential for subsequent bimolecular reactions – can be strongly impacted.
Full citation: Thøgersen, J., Bregnhøj, M., Weidner, T. and Jensen, F., The primary deep-UV photochemistry of aqueous fumarate and maleate, Phys. Chem. Chem. Phys., 28, 2965-2975 (2026). https://doi.org/10.1039/D5CP03064A
Lee Tiszenkel, Astrid N. Pedersen, Vignesh Vasudevan-Geetha, Margarete C. Hopf, Marianne Glasius, Jonas Elm, and Shan-Hu Lee
Aerosol nucleation accounts for the majority of secondary aerosols, yet it is unclear how biogenic and anthropogenic chemical precursors contribute to nucleation in mixed atmospheric environments. Here, we show laboratory experiments and quantum calculations, which demonstrate, for the first time to our knowledge, that highly oxygenated organosulfates (OOS) formed in the gas phase contribute to aerosol nucleation independently of sulfuric acid and oxygenated organic molecules. More than 200 different gas-phase OOS were detected with the nitrate CI-APi-TOF from a mixture of α-pinene, ozone, and SO2. The gas-phase OOS concentrations were strongly correlated with sulfuric acid. Quantum chemical modeling simulations showed that OOS forms in the gas phase from α-pinene diols and SO3 via a barrierless process. Nucleation rates increased much more rapidly with increasing α-pinene concentrations in the mixed system than in the pure biogenic system, clearly demonstrating that OOS are effective nucleation precursors in mixed biogenic and anthropogenic systems.
Full citation: , , , , , , & Oxygenated organosulfates are an effective nucleation precursor in mixed biogenic and anthropogenic environments, Geophysical Research Letters, 53, e2025GL117259, (2026). https://doi.org/10.1029/2025GL117259
Freja Hasager, Þuríður Nótt Björgvinsdóttir, Michele Curzel, and Marianne Glasius
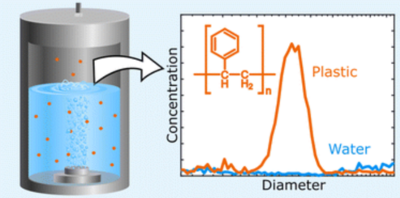
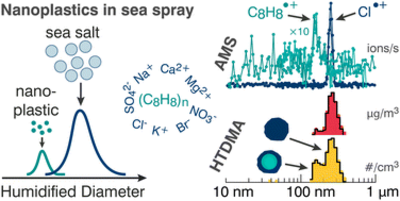
There is a need to develop analytical techniques for detection of airborne microplastic particles due to increasing evidence of their presence in ambient air and the associated possible health and climate effects. Many current analytical methods are limited by particle size and subjective visual evaluation. We have tested and validated an analytical pyrolysis gas chromatography-mass spectrometry, py-GC-MS, method that was developed specifically for detection of micro- and nanoplastic particles in aerosol samples. Some of the most common polymers, namely polyethylene (PE), polypropylene (PP), polyvinyl chloride (PVC), polyethylene terephthalate (PET), polyurethane (PU), polyamide (PA) and polystyrene (PS), were mixed and analyzed in atmospherically relevant sample matrices. A temperature-ramped selected ion monitoring (SIM) method was developed to detect them, based on characteristic pyrolysis products identified in pyrograms of each polymer individually and in their mixtures. Calibration curves in the range 10 ng – 1 µg were determined for each polymer with R2 values ranging from 0.72 to 0.99. A negative matrix effect was observed for reactive pyrolysis products, which was stronger in samples containing more aerosol matrix. This effect may be caused by secondary reactions between the pyrolysis products and aerosol matrix components. On the contrary a positive matrix effect was observed for PVC, possibly due to the addition of alkenes by the matrix which partake in radical reactions to form naphthalene. Deuterated polystyrene was tested as an internal standard to account for the matrix effect, but it was found to only be suitable for PS and PP. Other internal standards are thus needed for the other polymers. This study presents a method for direct analysis of aerosol particle samples with quantification of PP, PS, and PA on the nanogram scale and semi-quantification of PE, PU, PET, and PVC, also in the sub-microgram range.
Full citation: Hasager, F., Björgvinsdóttir, Þ. N., Curzel, M. and Glasius, M., Development and validation of an analytical pyrolysis method for determination of microplastic polymers in atmospheric aerosols, Journal of Analytical and Applied Pyrolysis, 193, 2, 107423, (2026). https://doi.org/10.1016/j.jaap.2025.107423
Alina Mostovaya, Lotte Dyrholm Thomsen, Mikael K. Sejr, Marianne Glasius, and Johnna M. Holding
Rapidly melting Arctic glaciers deliver increasing amounts of allochthonous material to the coastal ocean, altering carbon cycling and promoting heterotrophy. As key factors influencing the activity of heterotrophic microbes, the quantity and quality of Arctic coastal organic carbon warrant closer examination. We investigated the molecular composition of dissolved organic matter (DOM) in two rivers and surface waters of Young Sound, NE Greenland—a high Arctic fjord where glacial runoff contributes to low primary productivity and increasing heterotrophy. Using ultra-high-performance liquid chromatography–tandem mass spectrometry (UHPLC-qTOF-MS), we conducted a non-targeted analysis of solid-phase extracted DOM (SPE-DOM). We expected DOM composition to differ between the two studied rivers (Tyroler and Zackenberg), which contrast in length, glacial water source, and catchment characteristics, and to reflect the salinity gradient in fjord waters. Both rivers carried a strong glacial imprint, with DOM enriched in aliphatic constituents typically associated with higher bioavailability, yet the proglacial Zackenberg River also exhibited unique compositional features that were more unsaturated and aromatic in character. Comparisons along a salinity gradient, from river plumes to outer fjord and open sea, revealed limited contrasts beyond the most glacially influenced section, with SPE-DOM composition showing high similarity across sites. Although multiple factors may contribute to this similarity, dilution and rapid processing of glacially derived DOM are likely to play a role. While further research is needed to understand carbon cycling in high Arctic fjords, our findings offer relevant insight into the molecular characteristics and potential ecological roles of DOM in this environment.
Full citation: , , , , & Molecular signatures of dissolved organic matter across the glacial, proglacial, and fjord continuum in NE Greenland. Journal of Geophysical Research: Biogeosciences, 130, e2025JG009161 (2025). https://doi.org/10.1029/2025JG009161
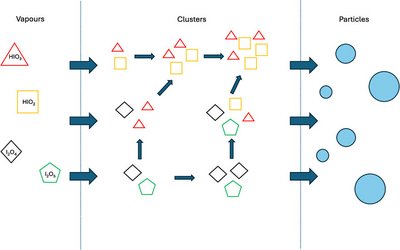
Morten Engsvang and Jonas Elm
Iodine-driven nucleation is thought to be a significant source of new particle formation, especially in marine and polar regions. Despite numerous studies, the mechanism is still not fully understood. To shed further light on this, we apply ZORA-DLPNO–CCSD(T0)/TZVPP//ωB97X-D3BJ/aug-cc-pVTZ-PP to calculate the thermochemistry of iodine-containing clusters up to tetramers and simulate the cluster formation potential for several nucleation paths using the atmospheric cluster dynamics code (ACDC). We find that iodine oxyacid–amine nucleation can be competitive with sulfuric acid–amine nucleation if iodic acid is present in a 10:1 ratio compared to sulfuric acid. Therefore, the importance of the iodine-driven pathway is regionally dependent. Likewise, we find that increasing the relative humidity from 34 to 73% only changes the cluster formation potential by a factor of 2. Nucleation pathways consisting of only iodic and iodous acid are unable to explain the relative nucleation rates previously observed in experiments. In contrast, the simultaneous nucleation of iodine oxides, assisted by iodine oxyacids, is better able to describe the trend. This indicates that a nucleation pathway starting with iodine oxides is more likely to be able to explain observed particle numbers. However, this current model does not include all of the hydrates of the clusters and does not account for the hydrolysis reactions of the iodine oxides. This would need to be incorporated in future studies.
Full citation: Engsvang, M. and Elm, J., Iodine Clusters in the Atmosphere II: Cluster Formation Potential of Iodine Oxyacids and Iodine Oxides, ACS Omega, 10 (23), 24887-24896 (2025). https://doi.org/10.1021/acsomega.5c02147
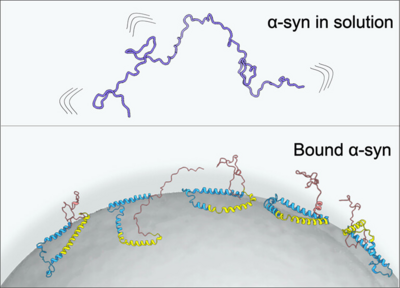
Akriti Mishra, Thaddeus W. Golbek, Asger Berg Thomassen, Lorena Zuzic, Lars Schmüser, Khezar Hayat Saeed, Fani Madzharova, Janni Nielsen, Birgit Schiøtt, Daniel E. Otzen, and Tobias Weidner
The impact of micro- and nanoplastics (MNPs) on human health is a growing field of research. Reports that MNPs can breach the blood-brain barrier and accumulate inside the brain have raised concerns over their possible involvement in the development of neurogenerative diseases. The aggregation of the abundant neuronal protein α-synuclein (α-syn) is pertinent to almost 50 neurological diseases including Parkinson’s disease (PD). The role of nanoplastics in the formation of toxic aggregates is unclear and has been shown to depend strongly on the type of plastics. Here we report the molecular structure and orientation of human α-syn adsorbed on polystyrene NPs using interface-specific sum frequency scattering (SFS) and structure-sensitive two-dimensional infrared (2D IR) spectroscopy. The SFS experimental data were compared with the calculated spectra of several thousands of α-syn conformations generated from molecular dynamics simulations. The SFS results reveal that α-syn folds on polystyrene nanoplastics, adopting a partly helical structure with the N-terminus and nonamyloid component regions directly bound on the polystyrene nanosurface, while the C terminus protrudes away from the polystyrene interface. 2D IR results suggest that the entire α-syn corona comprises of partly aggregated α-syn structures, built of an ordered core enclosed with flexible dynamic regions. The data shed light on the mechanism by which α-syn folds and forms aggregates at the plastic particle surfaces, a link that has been missing in understanding the role of nanoplastic in the pathogenesis of PD and related neurodegenerative diseases.
Full citation: Mishra, A., Golbek, T. W., Thomassen, A. B., Zuzic, L., Schmüser, L., Saeed, K. H., Madzharova, F., Nielsen, J., Schiøtt, B., Otzen, D. E. and Weidner, T., Pathological Folding of α-Synuclein on Polystyrene Nanoplastic Revealed by Sum Frequency Scattering and 2D Infrared Spectroscopy, The Journal of Physical Chemistry Letters, 16, 45, 11893-11900, (2025). https://doi.org/10.1021/acs.jpclett.5c02526
Khezar H. Saeed, Sigurd F. Truelsen, and Tobias Weidner
Protein glycosylation is known to impact structural and functional dynamics, yet its influence on interfacial behavior remains underexplored. Here, we systematically investigate the effects of glycosylation on the binding orientation of the Thermomyces lanuginosus lipase (TLL) variants at the air/water interface. Using a combination of experimental vibrational sum frequency generation (VSFG) spectroscopy and spectral calculations, we directly probe the interfacial conformation of TLL with varying degrees of glycosylation. Our findings reveal that the lid-open conformation is preferred for both glycosylated and deglycosylated forms and that the N33Q point mutation does not significantly alter binding. Additionally, high-mannose glycosylation broadens the range of preferred orientations. Complementary surface pressure measurements show similar protein concentrations across variants, suggesting that the reduced VSFG intensity for glycosylated TLL arises from an increased interfacial disorder. These results demonstrate that glycosylation can indirectly modulate protein surface interactions, suggesting a broader role for this common post-translational modification in protein interfacial binding.
Full citation: Saeed, K. H., Truelsen, S. F., and Weidner, T., Glycosylation as a Facile Route to Control Enzyme Orientation at Interfaces, The Journal of Physical Chemistry Letters, 16, 46, 11964-11969, (2025). https://doi.org/10.1021/acs.jpclett.5c02420
Ásmundur Smári Ragnarsson, Irati Lasa Uriarte, Caitlin Howell, Khezar Hayat Saeed, and Tobias Weidner
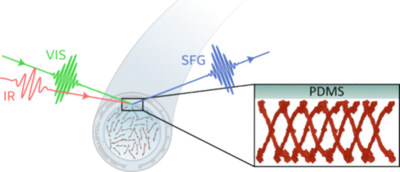
The adsorption of fibrinogen on biomaterial surfaces, particularly polydimethylsiloxane (PDMS), plays a key role in foreign body reactions and has recently been shown to be one of the main factors driving catheter-associated urinary tract infections (CAUTIs). Yet, despite detailed studies on the fibrinogen’s solution and crystal structures, its behavior at material interfaces is less understood. Using sum frequency generation (SFG) spectroscopy and structural modeling, we determined the binding pose and conformation of human fibrinogen at the PDMS-buffer interface. Fibrinogen adopts an upright orientation on PDMS with minimal bending. Comparisons with spectra recorded at the air–water interface and literature data on polystyrene reveal significant differences in orientation: fibrinogen binds flat and bent at these model hydrophobic interfaces, while upright conformations are observed on liquid PDMS. These findings demonstrate specific interaction beyond simple hydrophobic attraction at the PDMS interface and hint at the critical role of surface chemistry in dictating fibrinogen’s interfacial structure and its implications for biomaterial design aimed at reducing foreign body reactions and CAUTIs.
Full citation: Ragnarsson, Á. S., Uriarte, I. L., Howell, C., Saeed, K. H. and Weidner, T., The Orientation of Human Fibrinogen at Biomedically Relevant Polydimethylsiloxane–Water Interfaces, Langmuir, 41, 19, 12089-12095 (2025). https://doi.org/10.1021/acs.langmuir.5c00581
Louise N. Jensen, Kasper Kristensen, Emil M. Iversen, Manjula R. Canagaratna, Pontus Roldin, and Merete Bilde
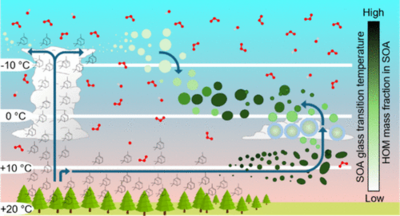
This study investigates how changes in temperature affect the secondary organic aerosol (SOA) phase state. SOA was formed by α-pinene ozonolysis in an atmospheric simulation chamber at temperatures (T0) in the range of 257–283 K at RH0 < 20%. After more than 14 h of SOA aging, one or more heating and cooling ramps were performed. Upon heating, we observe that the onset of evaporative SOA shrinkage is delayed by up to ∼20 K relative to T0. Our observations are supported by aerosol dynamics and kinetic multilayer model simulations, relating observed changes to an effectively reversible temperature- and SOA-composition-dependent phase transition from solid glassy to semisolid. We demonstrate that the SOA content of highly oxygenated organic molecules (HOMs) increases with T0 and at lower α-pinene concentrations. Higher HOM SOA content results in more viscous SOA with a higher glass transition temperature (Tg). The model was used to quantify how Tg varies with T0 and the amount of α-pinene being oxidized. Because the SOA phase state is influenced by the conditions under which it forms, and affects SOA lifetime, reactivity, water uptake, and potentially ice nucleating properties, the results presented herein may have wide implications for the design of future SOA experiments, air quality, and climate.
Full citation: Jensen, L. N., Kristensen, K., Iversen, E. M., Canagaratna, M. R., Roldin, P., and Bilde, M., Nature of Temperature-Induced Phase Transitions in Secondary Organic Aerosol Particles, Environmental Science & Technology, 59, 45, 24359–24367, (2025). https://doi.org/10.1021/acs.est.5c08582
Christian D. F. Castenschiold, Claudia Mignani, Sigurd Christiansen, Malin Alsved, Luisa Ickes, Sylvie V. M. Tesson, Jakob Löndahl, Merete Bilde, Thomas Bataillon, Kai Finster, and Tina Šantl-Temkiv
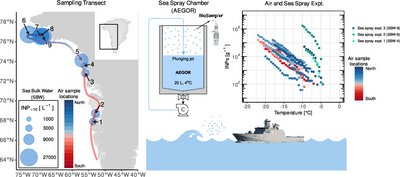
Biogenic ice-nucleating particles (INPs) can significantly impact mixed-phase clouds by enhancing precipitation and reducing albedo. As Arctic sea ice diminishes, the exposure of open ocean may increase aerosolization rates of marine bioaerosols and INPs. We investigated INP concentrations and microbial communities in ambient marine air, sea bulk water (SBW), and sea surface microlayer (SML) along a transect from the Davis Strait to Baffin Bay. INP concentrations in SBW increased with latitude, regardless of the extent of terrestrial freshwater input. We further identified correlations between INP levels and abundances of specific microbial taxa, including Formosa, Lewinella, Micromonas, and Dino-Group-I-Clade-5, suggesting potential ice nucleation activity of these taxa. Air samples exhibited distinct microbiomes compared to seawater, indicating terrestrial contributions, but at the highest observed wind speeds (7–8 m/s), substantial contributions of the seawater microbiome were detected in the air. Elevated atmospheric INP concentrations at higher latitudes correlated with seawater INP levels, which was supported by laboratory sea spray experiments showing that INPs in SBW influenced aerosol INP levels. Our findings highlight the Arctic Ocean as a significant source of biogenic atmospheric INPs and enhance our understanding of marine microbes as contributors to biogenic INPs. By identification of potential ice nucleation active microbial taxa and examination of aerosolization processes, this study provides a framework for future research on Arctic marine-derived INPs and their atmospheric impact.
Full citation: Castenschiold, C. D. F., Mignani, C., Christiansen, S., Alsved, M., Ickes, L., Tesson, S. V. M., Löndahl, J., Bilde, M., Bataillon, T., Finster, K., and Šantl-Temkiv, T., Nature of Temperature-Induced Phase Transitions in Secondary Organic Aerosol Particles, Environmental Science & Technology, 59, 42, 22518–22532, (2025). https://doi.org/10.1021/acs.est.5c03650
Fabian Mahrt, Sepehr Nikkho, Julia Zaks, Gurcharan Uppal, Anita Lam, Markus Ammann, and Allan K. Bertram
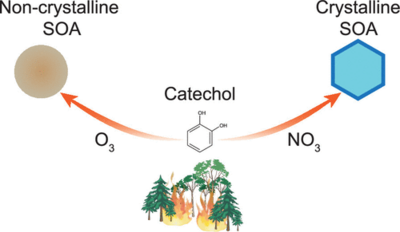
Biomass burning events, including wildfires, emit large amounts of phenolic compounds such as catechol. These compounds can react with nitrate radicals (NO3), a key nighttime oxidant, to form secondary organic aerosol (SOA). Although SOA is traditionally assumed to be noncrystalline, we present surprising evidence from X-ray diffraction that SOA formed from catechol + NO3 in an atmospheric simulation chamber contains crystalline material. In addition, the diffraction pattern and mass spectrum of this SOA closely resemble those of nebulized crystalline 4-nitrocatechol (4-NC), suggesting the presence of crystalline 4-NC within the SOA. These findings help explain unusual size distributions of catechol + NO3 SOA observed in prior studies and conflicting measurements of 4-NC’s effective saturation vapor concentrations. Calculations of 4-NC’s melting temperature as a function of its mole fraction in SOA, combined with observations of ambient 4-NC concentrations, suggest that 4-NC can exist in a solid crystalline phase state at temperatures below 288 K in wildfire plumes in the atmosphere. The presence of crystalline 4-NC and crystalline SOA in wildfire plumes may affect particle size distributions, cloud formation, and heterogeneous and photolytic reaction rates, with potentially important implications for atmospheric chemistry, air quality, and climate, warranting additional studies on this topic.
Full citation: Mahrt, F., Nikkho, F., Zaks, J., Uppal, G., Lam, A., Ammann, M., and Bertram, A. K., Surprising Crystallinity of Biomass Burning Secondary Organic Aerosol from Catechol and Nitrate Radical Reactions: Evidence and Possible Implications, Environmental Science & Technology, 59, 32, 16923-16932, (2025). https://doi.org/10.1021/acs.est.5c06834
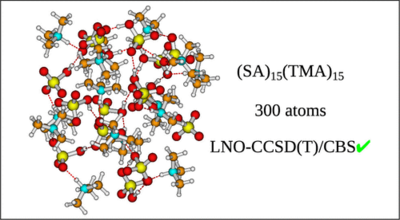
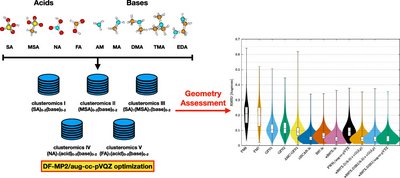
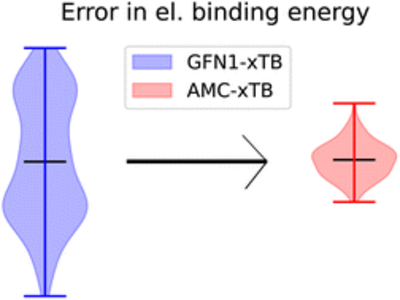
Yosef Knattrup and Jonas Elm
Aerosols are the largest source of uncertainty in modern global radiative forcing modeling. Atmospheric molecular clusters are important intermediates in atmospheric new particle formation (NPF). The evaporation rate of clusters can be calculated using quantum chemical methods, with an exponential dependence on the free energy. Hence, for simulating accurate NPF rates, high-accuracy calculations are needed. We have constructed a versatile benchmark set of 218 conformers of atmospheric molecular dimer clusters consisting of sulfuric acid (SA), formic acid (FA), nitric acid (NA), methanesulfonic acid (MSA), water (W), ammonia (AM), methylamine (MA), dimethylamine (DMA), trimethylamine (TMA), and ethylenediamine (EDA) molecules. Using this test set, we benchmark the local coupled cluster methods, DLPNO–CCSD(T0) and LNO–CCSD(T), using different basis sets and locality settings, and test extrapolation procedures to the complete basis set (CBS), local approximation free (LAF), and complete PNO space (CPS) limits. The extrapolations are tested against the binding energies of high-level CCSD(F12*)(T+)/cc-pVTZ-F12 reference calculations. We find that the LNO–CCSD(T) methods offer a better accuracy-to-cost ratio for atmospheric molecular clusters than the usually employed DLPNO–CCSD(T0) method. Furthermore, the CBS limit extrapolation using the aug-cc-pVTZ and aug-cc-pVQZ basis sets should be readily attainable for the LNO–CCSD(T) method on the usually studied cluster sizes (4–8 monomers). Simulating the new particle formation rate of the (SA)1–4(AM)1–4 and (SA)1–4(DMA)1–4 systems using the Atmospheric Cluster Dynamics Code, we find an increased sensitivity to the locality settings for larger clusters, but the basis set error is still the most dominant. Hence, simulated cluster formation rates would also benefit from doing LAF extrapolation. Finally, we illustrate the calculations of LNO–CCSD(T)/CBS binding energies of a large (SA)15(TMA)15 cluster (300 atoms). Hence, the application of LNO–CCSD(T) allows for significantly more accurate binding energies of much larger clusters than previously possible.
Full citation: Knattrup, Y. and Elm, J., Extrapolating Local Coupled Cluster Calculations toward CCSD(T)/CBS Binding Energies of Atmospheric Molecular Clusters, ACS Omega, 10, 40, 46794-46808, (2025). https://doi.org/10.1021/acsomega.5c04476
Tim Steinert, David Ginsbourger, August Lykke-Møller, Ove Christiansen, and Henry Moss
We study the incorporation of equivariances into vector-valued GPs and more general classes of random field models. While kernels guaranteeing equivariances have been investigated previously, their evaluation is often computationally prohibitive due to required integrations over the involved groups. In this work, we provide a kernel characterization of stochastic equivariance for centred second-order vector-valued random fields and we construct integration-free equivariant kernels based on the notion of fundamental regions of group actions. We establish data-efficient and computationally lightweight GP models for velocity fields and molecular electric dipole moments and demonstrate that proposed integration-free kernels may also be leveraged to extract equivariant components from data.
Full citation: Steinert, T., Ginsbourger, D., Lykke-Møller, A., Christiansen, O. and Moss, H., Integration-free kernels for equivariant Gaussian process modelling, Proceedings: 42nd International Conference on Machine Learning, (2025). https://openreview.net/forum?id=hYxZJycvrz
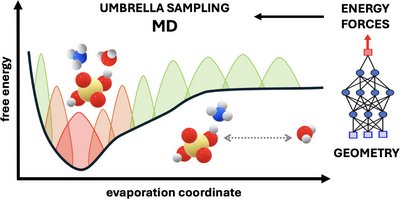
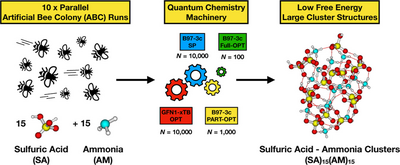
Jakub Kubečka, Yosef Knattrup, Georg Baadsgaard Trolle, Bernhard Reischl, August Smart Lykke-Møller, Jonas Elm, and Ivo Neefjes
Accurately modeling the binding free energies associated with molecular cluster formation is critical for understanding atmospheric new particle formation. Conventional quantum-chemistry methods, however, often struggle to describe thermodynamic contributions, particularly in systems exhibiting significant anharmonicity and configurational complexity. We employed umbrella sampling, an enhanced-sampling molecular dynamics technique, to compute Gibbs binding free energies for clusters formed from a diverse set of new particle formation precursors, including sulfuric acid, ammonia, dimethylamine, and water. By performing umbrella sampling along the evaporation coordinate, using forces computed at the semiempirical GFN1-xTB level of theory, we effectively capture entropic effects such as vibrational anharmonicities and transitions between different configurational minima, while avoiding errors from symmetry overcounting. In addition, we explored machine-learning-enhanced umbrella sampling simulations using neural network potentials trained on higher-level quantum chemistry data, demonstrating the feasibility of this approach for improving accuracy while maintaining computational efficiency. Our results show improved agreement with experimental values compared to conventional methods. We also present examples of gas-to-particle uptake processes, providing insights into cluster and aerosol–surface chemistry using first-principles approaches rather than commonly used molecular-mechanics force fields. This study demonstrates the importance of accounting for dynamics in predicting molecular binding thermodynamics in complex environments and highlights the potential of combining physics-based simulations with machine learning for reliable and scalable predictions.
Full citation: Kubečka, J., Knattrup, Y., Trolle, G. B., Reischl, B., Lykke-Møller, A. S., Elm, J., and Neefjes, I., Thermodynamics of Molecular Binding and Clustering in the Atmosphere Revealed through Conventional and ML-Enhanced Umbrella Sampling, ACS Omega, 10 (34), 39148-39161, (2025). https://doi.org/10.1021/acsomega.5c05634
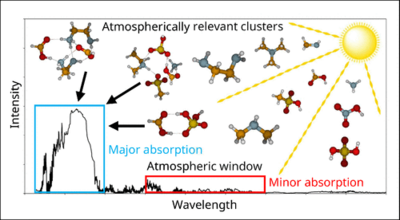
Georg Baadsgaard Trolle, Jakub Kubečka, and Jonas Elm
Information about the optical properties of atmospheric molecular clusters is scarce as they are challenging to measure using current experimental techniques. Here we explore the absorption and Rayleigh scattering properties of acid–base molecular clusters using quantum chemical methods. We studied 127 small (acid)1–2(base)1–2 cluster systems, with the acids sulfuric acid (SA), methanesulfonic acid (MSA), nitric acid (NA), and formic acid (FA) in all combinations of the bases ammonia (AM), methylamine (MA), dimethylamine (DMA), trimethylamine (TMA), and ethylenediamine (EDA). To further explore the effect of cluster size on the optical properties, we studied the large (SA)n(AM)n cluster systems, with n up to 15 acid–base pairs. We calculated the polarizability tensors and the 10 lowest excitation energies at the CAM-B3LYP/aug-cc-pVTZ level of theory. We find that the isotropic polarizability is almost linearly dependent on the cluster size, with small variations depending on the cluster composition. The anisotropic polarizability is plateauing as a function of cluster size. The larger the cluster, the more dominant the isotropic contribution becomes in the calculation of the Rayleigh light scattering activity. As a consequence, the Rayleigh scattering activity will increase quadratically as a function of cluster size. We stress that future studies on the scattering properties should be evaluated as effective scattering, taking the concentrations of the clusters into account. We find that the clusters absorb infrared (IR) radiation in the atmospheric spectral window region but speculate that their lifetime is too short to be competitive with common greenhouse gases. Due to the lack of strong chromophores in the studied acid–base clusters, the ultraviolet–visual (UV–vis) absorption is found to occur in the deep UV. Hence, clusters with more organic content should be studied in the future. Finally, we outline several directions in which the field of studying the optical properties of clusters and aerosols using response theory methods could evolve.
Full citation: Trolle, G. B., Kubečka, J., and Elm, J., Absorption and Scattering Properties of Atmospheric Molecular Clusters, The Journal of Physical Chemistry A, 129, 40, 9129-9138, (2025). https://doi.org/10.1021/acs.jpca.5c03658
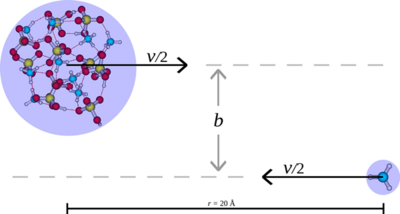
Yosef Knattrup, Ivo Neefjes, Jakub Kubečka, and Jonas Elm
When simulating new particle formation rates, collisions in the system are approximated as hard spheres without long-range interactions. This simplification may lead to an underestimation of the actual formation rate. In this study, we employ semi-empirical molecular dynamics (SEMD) at the GFN1-xTB level of theory to probe the sticking process of the monomers sulfuric acid (SA), methanesulfonic acid (MSA), nitric acid (NA), formic acid (FA), ammonia (AM), methylamine (MA), dimethylamine (DMA), and trimethylamine (TMA) onto freshly nucleated particles (FNPs). The FNPs considered are (SA)10(AM)10, (SA)10(MA)10, (SA)10(DMA)10, and (SA)10(TMA)10.
In general, we find that the hard-sphere kinetic approximation, which neglects long-range interactions, significantly underestimates the number of collisions leading to sticking. By calculating the sticking coefficient from SEMD simulations, we obtain enhancement factors of 2.3 and 1.5 for the SA + (SA)10(AM)10 and AM + (SA)10(AM)10 collisions, respectively. A comparison with OPLS (optimized potentials for liquid simulations) all-atom force field simulations shows similar enhancement factors of 2.4 and 1.6 for the SA + (SA)10(AM)10 and AM + (SA)10(AM)10 collisions, respectively.
Compared to the force field simulations, SEMD exhibits a more isotropic sticking behavior, with the probability remaining near unity for small offsets before rapidly dropping to 0 % beyond a certain offset. In contrast, the force field simulations show a more gradual decline in sticking probability due to certain orientations still leading to sticking. The largest discrepancy between the two methods occurs at lower collision velocities – below 200 m s−1 for SA and below 400 m s−1 for AM – where force field simulations, even for head-on collisions, predict low or zero sticking probability. This has previously been attributed to periodic repulsions between the rotating collision partners caused by fluctuations in their charge distributions. In contrast, SEMD simulations do not exhibit this behavior. Since these low velocities are not significantly populated in our simulations, both methods yield similar enhancement factors. However, for systems with larger effective masses, where such velocities are more prevalent, we would expect the two methods to diverge.
Full citation: Knattrup, Y., Neefjes, I., Kubečka, J., and Elm, J., Growth of atmospheric freshly nucleated particles: a semi-empirical molecular dynamics study, Aerosol Research, 3, 237–251, (2025), https://doi.org/10.5194/ar-3-237-2025.
Jan Thøgersen, Akriti Mishra, Tobias Weidner, and Frank Jensen
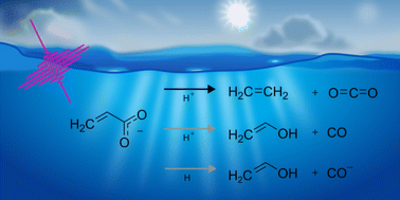
We apply transient absorption spectroscopy supported by 2D-IR spectroscopy and density functional theory calculations to determine the primary photolysis of acrylate excited via the 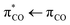 transition at 200 nm. Upon photoexcitation, about half of the excited acrylate anions return to the ground state and relax to equilibrium in 5 ps primarily through intermolecular coupling between the carboxylate group and the surrounding water. The rest of the excited acrylate anions dissociate. Three dissociation channels have been identified. In one reaction, decarboxylation of acrylate forms CO2 and CH2CH−. CH2CH− is protonated by water and forms ethene, C2H4, in <0.8 ps. In the second reaction, the excited acrylate anions dissociate to H2C
transition at 200 nm. Upon photoexcitation, about half of the excited acrylate anions return to the ground state and relax to equilibrium in 5 ps primarily through intermolecular coupling between the carboxylate group and the surrounding water. The rest of the excited acrylate anions dissociate. Three dissociation channels have been identified. In one reaction, decarboxylation of acrylate forms CO2 and CH2CH−. CH2CH− is protonated by water and forms ethene, C2H4, in <0.8 ps. In the second reaction, the excited acrylate anions dissociate to H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHO− and CO. In about 20 ps, H2CCHO− picks up a proton from water to produce vinyl alcohol, H2CCHOH. A third dissociation channel forms H2CCHO˙ and CO−. H2CCHO˙ abstracts a hydrogen atom from water and forms vinyl alcohol. Vinyl alcohol will tautomerize to acetaldehyde, but this occurs on a time scale longer than the experimental observation time of 0.56 ns.
CHO− and CO. In about 20 ps, H2CCHO− picks up a proton from water to produce vinyl alcohol, H2CCHOH. A third dissociation channel forms H2CCHO˙ and CO−. H2CCHO˙ abstracts a hydrogen atom from water and forms vinyl alcohol. Vinyl alcohol will tautomerize to acetaldehyde, but this occurs on a time scale longer than the experimental observation time of 0.56 ns.
Full citation: Thøgersen, J., Mishra, A., Weidner, T. and Jensen, F., The primary photolysis of aqueous acrylate, Physical Chemistry Chemical Physics, 27, 21297-21306, (2025), https://doi.org/10.1039/D5CP03023D.
Lars Henrik Olsen, Carolin König, Ove Christiansen
Based on a theoretical analysis of systems composed of subsystems described using a coupled cluster parametrization, we developed a vibrational coupled cluster embedding theory specifically tailored for the computation of response properties. This work identifies several strategies for calculating excitation energies, transition probabilities, and other response functions in large systems of interacting subsystems. A particularly effective embedding approach was formulated around a Lagrangian with multilinear interaction terms, yielding a structure that is nonlinear in both coupled cluster amplitudes and multipliers. Within this framework, we derived the corresponding response functions and associated eigenvalue equations. We also explored partitioning strategies for these equations, resulting in approximate exciton-like models that combine computational efficiency with conceptual clarity. This exciton-inspired methodology establishes a unified framework for computing excitation energies and transition properties in both electronic and vibrational coupled cluster response theories, applicable in both vacuum and embedded contexts. It provides a theoretical foundation for the future development of efficient methods for simulating vibrational spectra in extended systems.
Full citation: Olsen, L. H., König C., and Christiansen, O., A Subsystem Perspective on Vibrational Coupled Cluster Response Theory, The Journal of Physical Chemistry A, 129, 37, 8699-8713, (2025), https://doi.org/10.1021/acs.jpca.5c03752.
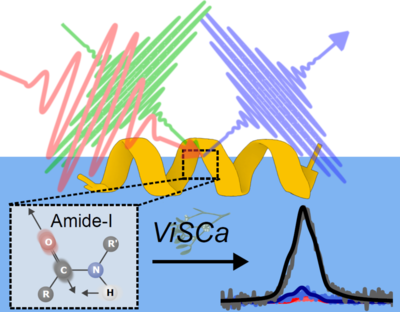
Khezar H. Saeed, Kris Strunge, Rolf Mertig, Steven J. Roeters, Tobias Weidner
Accurate spectral calculations are essential for interpreting the complex vibrational spectra of proteins, but high-level quantum chemical methods are computationally prohibitive for large proteins. This tutorial introduces the Vibrational Spectra Calculation (ViSCa) toolbox, a computational suite of methods for sum frequency generation (VSFG) spectral calculations. This article guides potential users through generating protein conformations and simulating the corresponding VSFG spectra to facilitate structural studies at interfaces. We use a frequency-mapping approach to construct an excitonic Hamiltonian of the amide-I modes of the protein backbone, which offers a functional balance between computational efficiency and accuracy. Included are several illustrative examples that showcase the different functionalities of ViSCa, including determining the orientation of a known protein structure at an interface (“ViSCa-Orient”), as well as coupling the calculations to molecular dynamics simulations to determine distinct changes in protein structure driven by the interfacial environment (“ViSCa-Select”).
Full citation: Saeed, K. H., Strunge, K., Mertig, R., Roeters, S. J., and Weidner, T., ViSCa: A computational toolbox for vibrational spectral calculations—Application to sum frequency generation spectroscopy of proteins, J. Chem. Phys., 163, 14, 144706, (2025). https://doi.org/10.1063/5.0296008
Sebastian Riis Thomsen, Nicolai Machholdt Høyer, Mads Greisen Højlund, and Ove Christiansen
The local bond-stretch (LBS) method is presented as a means of obtaining a set of localized, rectilinear vibrational modes. Three variants of the LBS method are considered: pure LBS, projected LBS (pLBS), and orthogonal, projected LBS (opLBS). These variants feature different degrees of localization and different coupling terms in the kinetic energy operator, such that the most localized method (LBS) has the largest number and magnitude of coupling terms, and the least localized (opLBS) has the least coupling terms. The different LBS variants are exemplified in computations on overtone vibrational spectra of water, nitroxyl (chemical formula HNO), formaldehyde, and 1,3-butadiene computed with a vibrational coupled cluster band Lanczos approach. These spectra are calculated using potential energy surfaces (PESs) obtained with the adaptive density-guided approach (ADGA). We observe faster convergence with respect to the coupling level in the PES when using the LBS variants compared to normal coordinates. Among the LBS variants, pLBS and opLBS appear most promising.
Full citation: Thomsen, S. R., Høyer, N. M., Højlund, M. G. and Christiansen, O., Local Bond-Stretch Coordinates for Anharmonic Vibrational Computations, The Journal of Physical Chemistry A, 129, 15, 3522-3536, (2025), https://doi.org/10.1021/acs.jpca.4c07704.
Zeynep Gündoğar, Mads Greisen Ho̷jlund, Kasper Green Larsen, and Ove Christiansen
We introduce an innovative recursive tensor decomposition method that draws inspiration from quantum chemical theories. This approach integrates ideas such as natural occupation numbers and natural basis, much like natural orbitals, and employs truncations that parallel the excitation-level truncations in the linear expansions of configuration interaction theory. The framework features recursive algorithms that combine linear expansion with natural basis transformations at each step, ensuring convergence to the original tensor. Consequently, a numerical technique is developed that reconstructs the initial tensor with precision within a predetermined tolerance, using only subtensors of limited dimension and a series of matrix transformations. An initial Python implementation has been created for the 3D tensor scenario where 3D tensors are decomposed to be represented using vectors and matrices alone. We illustrate the behavior of the final Recursive Linear Tensor Expansion in Natural basis algorithm in processing random data sets, experimental data sets from diverse sources with both real and complex tensors, and data sets representing both time-independent and time-dependent anharmonic vibrational wave functions of water. Finally, the systematic accuracy control is illustrated for density fitting two-electron repulsion integrals and tested for the second-order correlation energy of molecular nitrogen and benzene.
Full citation: Gündoğar, Z., Højlund, M. G., Larsen, K. G. and Christiansen, O., Recursive Linear Tensor Expansion with Natural Occupation Analysis, Journal of Chemical Theory and Computation, 21, 19, 9270-9289, (2025), https://doi.org/10.1021/acs.jctc.5c01101.
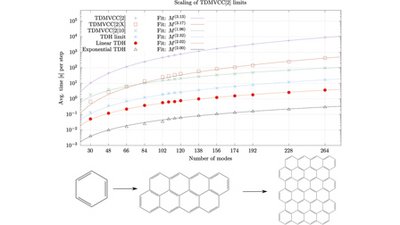
Mads Greisen Højlund, Alberto Zoccante, Andreas Buchgraitz Jensen, and Ove Christiansen
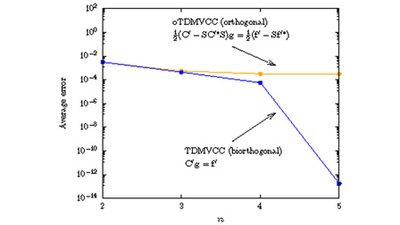
In recent decades, coupled cluster theory has proven valuable in accurately describing correlation in many-body systems, particularly in time-independent computations of molecular electronic structure and vibrations. This review describes recent advancements in using coupled cluster parameterizations for time-dependent wave functions for the efficient computation of the quantum dynamics associated with the motion of nuclei. It covers time-dependent vibrational coupled cluster (TDVCC) and time-dependent modal vibrational coupled cluster (TDMVCC), which employ static and adaptive basis sets, respectively. We discuss the theoretical foundation, including many-mode second quantization, bivariational principles, and various parameterizations of time-dependent bases. Additionally, we highlight key features that make TDMVCC promising for future quantum dynamical simulations. These features include fast configuration-space convergence, the use of a compact adaptive basis set, and the possibility of efficient implementations with a computational cost that scales only polynomially with system size.
Full citation: Højlund, M. G., Zoccante, A., Jensen, A. B., and Christiansen, O., Time-Dependent Vibrational Coupled Cluster Theory With Static and Dynamic Basis Functions, WIREs Computational Molecular Science, 15, e70001, (2025), https://doi.org/10.1002/wcms.70001.
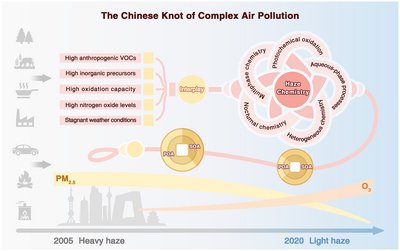
Ru-Jin Huang, Yong Jie Li, Qi Chen, Yanli Zhang, Chunshui Lin, Chak K. Chan, Jian Zhen Yu, Joost de Gouw, Shengrui Tong, Jingkun Jiang, Weigang Wang, Xiang Ding, Xinming Wang, Maofa Ge, Weijian Zhou, Doug Worsnop, Michael Boy, Merete Bilde, Ulrike Dusek, Annmarie G. Carlton, Thorsten Hoffmann, V. Faye McNeill, and Marianne Glasius
Air pollution is a global environmental problem with far-reaching implications for air quality, climate, ecosystems, and human health. Over the recent decades, China has experienced severe haze pollution, also called “complex air pollution.” These haze extremes are characterized by high concentrations of fine particulate matter (smaller than 2.5 μm, or PM2.5) and occur with extensive temporal and spatial coverage, although the situation was alleviated after 2013, owing to regulatory efforts. Distinct from the historical London fog caused mainly by coal combustion and the Los Angeles smog mainly formed by the photochemical oxidation of vehicular exhaust, haze pollution in China stems from high primary emissions and efficient secondary formation. One distinct feature of China at the current stage is a blend of agricultural and industrial societies, leading to high emissions of secondary aerosol precursors from diverse sources. These include NH3 from agricultural activities and anthropogenic volatile organic compounds (VOCs) from residential coal combustion and straw burning for heating and cooking (features of an agricultural society), as well as NOx (NO + NO2), SO2, and anthropogenic VOCs from vehicular exhaust and industrial activities (features of an industrial society). The mixture of these abundant inorganic and organic precursors can largely change the yields, chemical speciation, and formation pathways of secondary organic aerosol (SOA).
Numerous studies have revealed that air pollution in urban China is not only of enormous magnitude but also represents a distinct chemical regime less commonly observed elsewhere. The efficient formation of secondary aerosol in winter is distinct, which to a large extent drives the haze formation, particularly in recent winters after large reduction of primary aerosol. Higher-than-expected atmospheric oxidizing capacity in winter China is a combined result of photolysis of elevated HONO, alkene ozonolysis in the presence of NOx, anthropogenic halogen radicals, and the O2/H2O-involved interfacial oxidation and hydrolysis processes. This represents a previously unidentified chemical regime to describe the explosive growth of SOA and secondary inorganic aerosol in complex air pollution. In addition, the elevated NOx levels can lead to the formation of a variety of nitrogen-containing multifunctional oxidation products. For example, secondary organic nitrate was reported to account for more than 40% of organic aerosol mass during haze events in urban China, which is important for SOA enhancement and altered physicochemical properties. Also, the formation of peroxyacyl nitrates may worsen the regional air pollution by prolonging the effective lifetime of peroxyl radicals. Moreover, the increasing fractional contribution of nitrate over sulfate in recent years results in enhanced aerosol liquid water content, promoting aqueous-phase SOA formation, as revealed through multiple field studies during humid haze events.
Despite considerable advances over the past decade, the precursors, formation, and transformation of SOA in urban China and their impacts on the radiative budget and human health are still very uncertain. Molecular-level speciation of SOA precursors and multigeneration products are essential to elucidate the formation and fate of SOA, and particular focus should be given to less-explored precursors, such as semivolatile and intermediate-volatility organic compounds and volatile chemical products. Noticeably, the sharp decrease of PM2.5 concentrations in urban China over the past decade, which has not been reported anywhere else, has led to a substantial increase of surface O3 concentrations, which facilitates the formation of SOA. Quantitative understanding of the unrecognized sources responsible for the enhanced atmospheric oxidizing capacity in winter urban China is therefore critical. With accurate parameterizations of the above aspects, an improved SOA simulation is expected. As for mitigation, diagnosing the O3-NOx-VOC sensitivity at the city or regional scale is essential for a cost-effective strategy to prioritize control measures on precursors that lead to both high SOA and high O3 concentrations.
Full citation: Huang, R.J., Li, Y. J., Chen, Q., Zhang, Y., Lin, C., Chan, C. K., Yu, J. Z., de Gouw, J., Tong, S., Jiang, J., Wang, W., Ding, X., Wang, X., Ge, M., Zhou, W., Worsnop, D., Boy, M., Bilde, M., Dusek U., Carlton, A. G., Hoffmann, T., McNeill, V. F., and Glasius, M., Secondary organic aerosol in urban China: A distinct chemical regime for air pollution studies, Science, 389, eadq2840, (2025), https://doi.org/10.1126/science.adq2840.
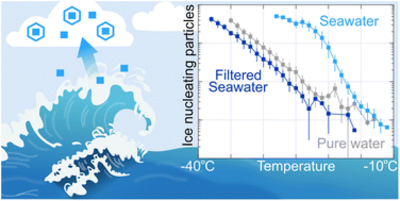
Eva R. Kjærgaard, Amanda S. Sejersen, Max F. Skov, Markus D. Petters, and Merete Bilde
Atmospheric ice nucleating particles (INPs) can affect cloud radiative properties and lifetimes and thus Earth's climate. Such particles may be emitted into the atmosphere from seawater via wave breaking processes. Here, we perform an exploratory investigation on the ice nucleating properties of seawater sampled on four days over a year (February, April, June, and November) from a coastal site near Aarhus in Denmark. We use a cold stage instrument (droplet size: 1 μL) to probe immersion mode freezing events. We find that bulk seawater contains INPs with T50 values around −20 °C independent of the month of sampling and INP concentrations ranging from 6 × 103 to 5 × 106 INP L−1 in a temperature range of −12 to −34 °C across all four samples. All samples displayed sensitivity to filtration (0.02 μm), as indicated by a decrease in INP concentration (lowering of freezing temperature). The filtered April and June samples froze at higher temperatures than the filtered November and February samples, which could indicate a variation in the population of INPs (>0.02 μm) over the year. Sea surface microlayer samples did not show enrichment of INPs compared to bulk seawater. Our results are discussed in the context of INP activity of seawater from other locations. While further studies are needed to understand the nature and potential seasonality of seawater INPs, we confirm the presence of INPs in coastal Baltic seawater that may contribute to atmospheric INP concentrations.
Full citation: Kjærgaard, E. R., Sejersen, A., Skov, M. F., Petters, M. D., and Bilde, M., Ice nucleating activity of coastal seawater from the entrance to the Baltic Sea, Environ. Sci.: Atmos., 5, 1014-1022, (2025), https://doi.org/10.1039/D5EA00031A
Galib Hasan, Haide Wu, Yosef Knattrup, Jonas Elm
Sulfuric acid (SA), ammonia (AM), and dimethylamine (DMA) are believed to be key contributors to new particle formation (NPF) in the atmosphere. NPF happens through gas-to-particle transformation via cluster formation. However, it is not obvious how small clusters grow to larger sizes and eventually form stable aerosol particles. Recent experimental measurements showed that the presence of mixtures of bases enhanced the nucleation rate by several orders of magnitude. Using quantum chemistry methods, this study explores this base synergy in the formation of large clusters from a mixture of SA, AM, and DMA. We calculated the binding free energies of the (SA)n(AM)x(DMA)n−x clusters, with n from 1 to 10, where x runs from 0 to n. The cluster structures were obtained using our recently developed comprehensive configurational sampling approach based on multiple ABCluster runs and meta-dynamic sampling via the Conformer–Rotamer Ensemble Sampling Tool (CREST). The structures and thermochemical parameters are calculated at the B97-3c level of theory. The final single point energy of the clusters is calculated at the ωB97X-D3BJ/6-311++G(3df,3pd) level of theory. Based on the calculated thermochemistry, we found that AM, despite being a weaker base, forms more intermolecular interactions than DMA and easily becomes embedded in the cluster core. This leads to the mixed SA–AM–DMA clusters being lower in free energy compared to the pure SA–AM and SA–DMA clusters. We find that the strong base DMA is important in the very first steps in cluster formation, but for larger clusters an increased ammonia content is found. We also observed that the cluster-to-particle transition point for the mixed SA–AM–DMA clusters occurs at a cluster size of 14 monomers, which is notably smaller than the transition points for the pure SA–AM (16 monomers) or pure SA–DMA (20 monomers) systems. This indicates a strong synergistic effect when both AM and DMA are present, leading to the formation of stable freshly nucleated particles (FNPs) at smaller cluster sizes. These findings emphasize the importance of considering several base molecules when studying the formation and growth of FNPs.
Full citation: Hasan, G., Wu, H., Knattrup, Y., Elm, J., Base synergy in freshly nucleated particles, Aerosol Res., 3, 101-111, (2025), https://doi.org/10.5194/ar-3-101-2025
Yosef Knattrup, Jonas Elm
Sulfuric acid, ammonia, and amines are believed to be key contributors to the initial steps in new particle formation in the atmosphere. However, other compounds such as organic compounds or nitric acid are believed to be important for further growth at larger sizes. In this study, we investigate the potential uptake of first-generation oxidation products from α-pinene (pinic and pinonic acid) and isoprene (trans-β-IEPOX, β4-ISPOOH, and β1-ISOPOOH), a potential highly oxidised molecule (HOM), formic acid, and nitric acid. The uptake is probed onto (SA)10(base)10 freshly nucleated particles (FNPs), where SA denotes sulfuric acid, and the bases are ammonia (AM), methylamine (MA), dimethylamine (DMA), or trimethylamine (TMA). The addition free energies were calculated at the ωB97X-D3BJ/6-311++G(3df,3pd)//B97-3c level of theory. We find favourable addition free energies of −8 to −10 kcal mol−1 for the HOM, pinic acid, and pinonic acid on the less sterically hindered (SA)10(AM)10 and (SA)10(MA)10 FNPs. This suggests that isoprene oxidation products do not contribute to the early growth of FNPs, but the α-pinene products do, in accordance with their expected volatilities. Calculating the second addition of a pinic acid molecule or pinonic acid molecule on the (SA)10(AM)10 FNPs, we find that pinic acid maintains its large addition free energy decrease due to its two carboxylic acid groups interacting with the other monomer, as well as the FNP. The pinonic-acid addition free energy drops to −3.9 kcal mol−1 due to the weak interactions between the FNP and its carbonyl group and the lack of monomer–monomer interactions. Calculating the addition free energy under realistic atmospheric conditions, we find that the FNPs studied are too small (1.4 nm) to support the growth of the studied uptake monomers. We find that the accretion product pinyl diaterpenylic ester (PDPE; C17H26O8) yields an addition free energy value of −17.1 kcal mol−1 . This suggests that PDPE can overcome the strong Kelvin effect of a 1.4 nm FNP and lead to spontaneous uptake under ambient conditions.
Full citation: Knattrup, Y., Elm, J., Uptake of organic vapours and nitric acid on atmospheric freshly nucleated particles, Aerosol Res, 3, 125-137, (2025), doi.org/10.5194/ar-3-125-2025
Georg Baadsgaard Trolle, Jakub Kubečka, Jonas Elm
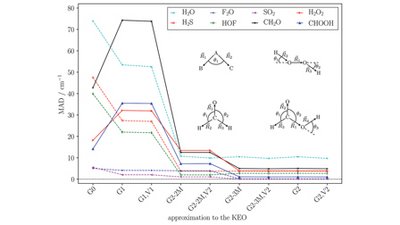
Pyruvic acid is an omnipresent compound in nature and is found both in the gas phase and in the particle phase of the atmosphere as well as in aqueous solution in the hydrosphere. Despite much literature on the photochemical degradation and stability of pyruvic acid in different chemical environments, the study of simultaneous interactions between gas-phase pyruvic acidor similar carboxylic acids with water and ions is not well-understood. Here, we present a study of microhydrated molecular clusters containing pyruvic acid and the structurally analogous carboxylic acids lactic acid, propionic acid, and 2,2-dihydroxypro-panoic acid by probing geometries, binding free energies, hydrate distributions, as well as their infrared (IR) absorption spectra. We performed a meticulous configurational sampling protocol for the various hydrated clusters ranging from low level of theory to high level of theory to identify the lowest free energy structure. We find that cluster geometries and especially their water structure are highly sensitive to the presence and character of ions. We show that the hydration of the studied organic acids is thermodynamically unfavorable in the gas phase and ions are necessary for mediating interactions between organic acids and water thus stabilizing the clusters. Finally, we find a clear correlation between decreasing pyruvic acid carboxylic O−H stretching frequencies, increasing intensity when adding more water to the clusters, and a correlation between increasing redshifting of the O−H frequencies upon addition of ions to the clusters. The observations done in this study could pave the way to unravel the mechanisms behind the transitioning of organic acids from the gas phase to the particle phase.
Full citation: Trolle, G. B., Kubečka, J., Elm, J., Modeling Local Aerosol Surface Environments: Clustering of Pyruvic Acid Analogs, Water, and Na+, Cl− Ions, ACS Omega, 10 (1), 1470-1485, (2025). https://doi.org/10.1021/acsomega.4c09196
Emil Mark Iversen, Merete Bilde, Henrik B.Pedersen
We describe three developments at the AURA atmospheric simulation chamber (made of Teflon and with a volume of ∼5 m3) aimed at an improved understanding of the physical conditions of the chamber to facilitate a better basis for comparisons between experimental data and results from numerical models. First, we demonstrate how the volume of the AURA chamber can be monitored by observing the position of a chamber wall using fixed laser distance sensors. The absolute volume calibration is obtained through a measurement of the relative humidity in the chamber during a controlled dilution experiment. Second, through a direct measurement, we characterize the occurrence, magnitude (∼0 – 80 kV/m), and decay time (∼10 – 20 h) of static electric fields inside the AURA chamber after charging. Further, we confirm directly that the AURA chamber can be significantly discharged and kept in a steady mode of charging with the addition of ion fans to the enclosure where the chamber is suspended. Third, we improve the air mixing capabilities at the AURA chamber by adding two mixing fans that allows efficient mixing of the chamber air within a few minutes. We characterize the effect of the electric field in the chamber and the rate of air mixing by direct measurements of the particle loss rate of injected polydisperse ammonium sulfate particles.
Full citation: Iversen, M. E., Bilde, M., Pedersen, B. H., Developments at the AURA atmospheric simulation chamber to characterize chamber volume, air mixing, and charging, Aerosol Science and Technology, 59 (5), 521–543, (2025). https://doi.org/10.1080/02786826.2024.2429658
Haide Wu, Yosef Knattrup, Andreas Buchgraitz Jensen, Jonas Elm
The formation of molecular clusters is an imperative step leading to the formation of new aerosol particles in the atmosphere. However, the point at which a given assembly of molecules represents an atmospheric molecular cluster or a particle remains ambiguous. Applying quantum chemical calculations, we elucidate this cluster-to-particle transition process in atmospherically relevant sulfuric acid–base clusters. We calculate accurate thermodynamic properties of large (SA)n(base)n clusters (n = 1–15), with SA being sulfuric acid and the base being either ammonia (AM), methylamine (MA), dimethylamine (DMA) or trimethylamine (TMA). Based on our results, we deduce property-based criteria for defining freshly nucleated particles (FNPs), which act as a boundary between discrete cluster configurations and large particles. We define the onset of FNPs as being when one or more ions are fully solvated inside the cluster and when the gradient of the size-averaged binding free energy approaches zero. This definition easily allows the identification of FNPs and is applicable to particles of arbitrary chemical composition. For the (SA)n(base)n clusters studied here, the cluster-to-particle transition point occurs around 16–20 monomers. We find that the formation of FNPs in the atmosphere depends greatly on the cluster composition and atmospheric conditions. For instance, at low temperature (278.15 K) and high precursor concentration (AM = 10 ppb and MA = 10 ppt), the SA–AM and SA–MA systems can form clusters that grow to and likely beyond ~1.8 nm sizes. The SA–DMA system forms clusters that grow to larger sizes at low temperature (278.15 K), independent of the concentration (DMA = 1–10 ppt), and the SA–TMA system (1 : 1 acid–base ratio) can only form small clusters that are unable to grow to larger sizes under the studied conditions.
Full citation:
Wu, H., Knattrup, Y., Jensen, A.B., Elm, J., Cluster-to-particle transition in atmospheric nanoclusters, Aerosol Res., 2, 303–314, (2024), https://doi.org/10.5194/ar-2-303-2024
Linjie Li, Ditte Thomsen, Cheng Wu, Michael Priestley, Emil Mark Iversen, Jane Tygesen Skønager, Yuanyuan Luo, Mikael Ehn, Pontus Roldin, Henrik B. Pedersen, Merete Bilde, Marianne Glasius, Mattias Hallquist
Formation of oxidized products from Δ3-carene (C 10H 16) ozonolysis and their gas-to-particle partitioning at three temperatures (0, 10, and 20 °C) under dry conditions (<2% RH) and also at 10 °C under humid (78% RH) conditions were studied using a time-of-flight chemical ionization mass spectrometer (ToF-CIMS) combined with a filter inlet for gases and aerosols (FIGAERO). The Δ3-carene ozonolysis products detected by the FIGAERO-ToF-CIMS were dominated by semivolatile organic compounds (SVOCs). The main effect of increasing temperature or RH on the product distribution was an increase in fragmentation of monomer compounds (from C 10 to C 7 compounds), potentially via alkoxy scission losing a C 3 group. The equilibrium partitioning coefficient estimated according to equilibrium partitioning theory shows that the measured SVOC products distribute more into the SOA phase as the temperature decreases from 20 to 10 and 0 °C and for most products as the RH increases from <2 to 78%. The temperature dependency of the saturation vapor pressure (above an assumed liquid state), derived from the partitioning method, also allows for a direct way to obtain enthalpy of vaporization for the detected species without accessibility of authentic standards of the pure substances. This method can provide physical properties, beneficial for, e.g., atmospheric modeling, of complex multifunctional oxidation products.
Full citation:
Li, L., Thomsen, D., Wu, C., Priestley, M., Iversen, M. E., Skønager, T. J., Luo, Y., Ehn, M., Roldin, P., Pedersen, B. H., Bilde, M., Glasius, M., Hallquist, M., Gas-to-Particle Partitioning of Products from Ozonolysis of Δ3-Carene and the Effect of Temperature and Relative Humidity, J. Phys. Chem. A, 128 (5), 918-928, (2024), https://doi.org/10.1021/acs.jpca.3c07316
Lærke Sloth Nielsen, Tina Šantl-Temkiv, María Palomeque Sánchez, Andreas Massling, Josephine Caroline Ward, Pia Bomholt Jensen, Thomas Boesen, Markus Petters, Kai Finster, Merete Bilde, Bernadette Rosati
Airborne microorganisms impact cloud formation and are involved in disease spreading. The ability of airborne cells to survive and express genes may be limited by reduced water availability in the atmosphere and depend on the ability of the cells to attract water vapor at subsaturated conditions, i.e., their hygroscopicity. We assessed hygroscopic properties of the plant pathogen Pseudomonas syringae, known to participate in cloud formation. We used a hygroscopicity tandem differential mobility analyzer to examine both hydration and dehydration behavior in the relative humidity (RH) range 5–90%. The cells were aerosolized either from Milli-Q water or from a 35 g L–1 NaCl solution, resulting in pure cells or cells associated with NaCl. Pure cells exhibited no deliquescence/efflorescence and a small gradual water uptake reaching a maximum growth factor (GF) of 1.09 ± 0.01 at 90% RH. For cells associated with NaCl, we observed deliquescence and a much larger maximum GF of 1.74 ± 0.03 at 90% RH. Deliquescence RH was comparable to that of pure NaCl, highlighting the major role of the salt associated with the cells. It remains to be investigated how the observed hygroscopic properties relate to survival, metabolic, and ice-nucleation activities of airborne P. syringae.
Full citation:
Nielsen, L. S., Šantl-Temkiv, T., Sánchez, M. P., Massling, A., Ward, J. C., Jensen, P. B., Boesen, T., Petters, M., Finster, K., Bilde, M., Rosati, B., Water Uptake of Airborne Cells of P. syringae Measured with a Hygroscopicity Tandem Differential Mobility Analyzer, Environ. Sci. Technol., 58 (43), 19211–19221, (2024), https://doi.org/10.1021/acs.est.4c01817
Yuanyuan Luo, Ditte Thomsen, Emil Mark Iversen, Pontus Roldin, Jane Tygesen Skønager, Linjie Li, Michael Priestley, Henrik B. Pedersen, Mattias Hallquist, Merete Bilde, Marianne Glasius, and Mikael Ehn
Δ3-carene is a prominent monoterpene in the atmosphere, contributing significantly to secondary organic aerosol (SOA) formation. However, knowledge about Δ3-carene oxidation pathways, particularly regarding their ability to form highly oxygenated organic molecules (HOMs), is still limited. In this study, we present HOM measurements during Δ3-carene ozonolysis under various conditions in two simulation chambers. We identified numerous HOMs (monomers: C7−10H10−18O6−14; dimers: C17−20H24−34O6−18) using a chemical ionization mass spectrometer (CIMS). Δ3-carene ozonolysis yielded higher HOM concentrations than α-pinene, with a distinct distribution, indicating differences in formation pathways. All HOM signals decreased considerably at lower temperatures, reducing the estimated molar HOM yield from ∼ 3 % at 20 °C to ∼ 0.5 % at 0 °C. Interestingly, the temperature change altered the HOM distribution, increasing the observed dimer-to-monomer ratios from roughly 0.8 at 20 °C to 1.5 at 0 °C. HOM monomers with six or seven O atoms condensed more efficiently onto particles at colder temperatures, while monomers with nine or more O atoms and all dimers condensed irreversibly even at 20 °C. Using the gas- and particle-phase chemistry kinetic multilayer model ADCHAM, we were also able to reproduce the experimentally observed HOM composition, yields, and temperature dependence.
Full citation:
Luo, Y., Thomsen, D., Iversen, E. M., Roldin, P., Skønager, J. T., Li, L., Priestley, M., Pedersen, H. B., Hallquist, M., Bilde, M., Glasius, M., and Ehn, M.: Formation and temperature dependence of highly oxygenated organic molecules (HOMs) from Δ3-carene ozonolysis, Atmos. Chem. Phys., 24, 9459–9473 (2024). doi.org/10.5194/acp-24-9459-2024
Andreas Buchgraitz Jensen and Jonas Elm
Atmospheric molecular clusters are important for the formation of new aerosol particles in the air. However, current experimental techniques are not able to yield direct insight into the cluster geometries. This implies that to date there is limited information about how accurately the applied computational methods depict the cluster structures. Here we massively benchmark the molecular geometries of atmospheric molecular clusters. We initially assessed how well different DF-MP2 approaches reproduce the geometries of 45 dimer clusters obtained at a high DF-CCSD(T)-F12b/cc-pVDZ-F12 level of theory. Based on the results, we find that the DF-MP2/aug-cc-pVQZ level of theory best resembles the DF-CCSD(T)-F12b/cc-pVDZ-F12 reference level. We subsequently optimized 1283 acid–base cluster structures (up to tetramers) at the DF-MP2/aug-cc-pVQZ level of theory and assessed how more approximate methods reproduce the geometries. Out of the tested semiempirical methods, we find that the newly parametrized atmospheric molecular cluster extended tight binding method (AMC-xTB) is most reliable for locating the correct lowest energy configuration and yields the lowest root mean square deviation (RMSD) compared to the reference level. In addition, we find that the DFT-3c methods show similar performance as the usually employed ωB97X-D/6-31++G(d,p) level of theory at a potentially reduced computational cost. This suggests that these methods could prove to be valuable for large-scale screening of cluster structures in the future.
Full citation:
Jensen, A. B., Elm, J., Massive Assessment of the Geometries of Atmospheric Molecular Clusters, J. Chem. Theory Comput., 20 (19), 8549–8558, (2024). https://doi.org/10.1021/acs.jctc.4c01046
Jakub Kubecka, Daniel Ayoubi, Zeyuan Tang, Yosef Knattrup, Morten Engsvang, Haide Wu and Jonas Elm
The computational cost of accurate quantum chemistry (QC) calculations of large molecular systems can often be unbearably high. Machine learning offers a lower computational cost compared to QC methods while maintaining their accuracy. In this study, we employ the polarizable atom interaction neural network (PaiNN) architecture to train and model the potential energy surface of molecular clusters relevant to atmospheric new particle formation, such as sulfuric acid–ammonia clusters. We compare the differences between PaiNN and previous kernel ridge regression modeling for the Clusteromics I–V data sets. We showcase three models capable of predicting electronic binding energies and interatomic forces with mean absolute errors of <0.3 kcal mol−1 and <0.2 kcal mol−1 Å−1 , respectively. Furthermore, we demonstrate that the error of the modeled properties remains below the chemical accuracy of 1 kcal mol−1 even for clusters vastly larger than those in the training database (up to (H2SO4)15(NH3)15 clusters, containing 30 molecules). Consequently, we emphasize the potential applications of these models for faster and more thorough configurational sampling and for boosting molecular dynamics studies of large atmospheric molecular clusters.
Full citation:
Kubecka, J., Ayoubi, D., Tang, Z., Knattrup, J., Engsvang, M., Wu, H., Elm, J., Accurate modeling of the potential energy surface of atmospheric molecular clusters boosted by neural networks, Environ. Sci.: Adv., 3 (10), 1438-1451 (2024), https://doi.org/10.1039/D4VA00255E
Eva R. Kjærgaard, Freja Hasager, Sarah S. Petters, Marianne Glasius, and Merete Bilde
Micro- and nanoplastic particles have been detected in most environmental compartments. The presence of microplastics in the remote marine atmosphere and close to large lakes suggests bubble mediated water–air transfer as a source of airborne microplastics, however, quantitative estimates of plastic emission from surface waters remain uncertain. In this work, we elucidate the emission of submicron polystyrene nanospheres by bubble bursting in a laboratory setting from low salinity waters (salinity 0–1.0 g kg−1), polystyrene particle diameter (103, 147 and 269 nm), aqueous particle number concentrations in the range 4 × 107–2 × 109 cm−3, and bubble formation rate (0.88–3.35 L min−1 of air). Production of polystyrene aerosols was demonstrated using a scanning mobility particle sizer and confirmed by analysis of filter samples using pyrolysis gas chromatography coupled to mass spectrometry. We show that production of polystyrene aerosol particles scales linearly with the number concentration of plastic particles in the water. Our results suggest that small amounts (0.01 g kg−1) of salt increase polystyrene particle production. To the best of our knowledge this is the first study of bubble mediated water–air transfer of plastic particles as small as 100 nm.
Full citation:
Kjærgaard, E. R., Hasager, F., Petters, S. S., Glasius, M., Bilde, M., Bubble-mediated generation of airborne nanoplastic particles, Environ. Sci.: Processes Impacts, 26 (7), 1216-1226 (2024). https://doi.org/10.1039/D4EM00124A
Mads Højlund and Ove Christiansen
We propose a new formulation of time-dependent coupled cluster with adaptive basis functions and division of the one-particle space into active and secondary subspaces. The formalism is fully bivariational in the sense of a real-valued time-dependent bivariational principle and converges to the complete-active-space solution, a property that is obtained by the use of biorthogonal basis functions. A key and distinguishing feature of the theory is that the active bra and ket functions span the same space by construction. This ensures numerical stability and is achieved by employing a split unitary/non-unitary basis set transformation: the unitary part changes the active space itself, while the non-unitary part transforms the active basis. The formulation covers vibrational as well as electron dynamics. Detailed equations of motion are derived and implemented in the context of vibrational dynamics, and the numerical behavior is studied and compared to related methods.
Højlund, M., Christiansen, O., A bivariational, stable, and convergent hierarchy for time-dependent coupled cluster with adaptive basis sets, J. Chem. Phys. 160 (17), 174119 (2024). https://doi.org/10.1063/5.0203914
Frederik Bader, David Lauvergnat, Ove Christiansen
Due to its efficiency and flexibility, the n-mode expansion is a frequently used tool for representing molecular potential energy surfaces in quantum chemical simulations. In this work, we investigate the performance of n-mode expansion-based models of kinetic energy operators in general polyspherical coordinate systems. In particular, we assess the operators with respect to accuracy in vibrationally correlated calculations and their effect on potential energy surface construction with the adaptive density guided approach. Our results show that the n-mode expansion-based operator variants are reliable and systematically improvable approximations of the full kinetic energy operator. Moreover, we introduce a workflow to generate the n-mode expanded kinetic energy operators on-the-fly within the adaptive density guided approach. This scheme can be applied in studies of species and coordinate systems, for which an analytical form of the kinetic energy operator is not available.
Full citation:
Bader, F., Lauvergnat, D., Christiansen, O., Efficient vibrationally correlated calculations using n-mode expansion-based kinetic energy operators, Phys. Chem. Chem. Phys., 26 (15), 11469-11481 (2024). https://doi.org/10.1039/D4CP00423J
Morten Engsvang, Haide Wu, and Jonas Elm
The contribution of iodine-containing compounds to atmospheric new particle formation is still not fully understood, but iodic acid and iodous acid are thought to be significant contributors. While several quantum chemical studies have been carried out on clusters containing iodine, there is no comprehensive benchmark study quantifying the accuracy of the applied methods. Here, we present the first study in a series that investigate the role of iodine species in atmospheric cluster formation. In this work, we have studied the iodic acid, iodous acid, iodine tetroxide, and iodine pentoxide monomers and their dimers formed with common atmospheric precursors.
We have tested the accuracy of commonly applied methods for calculating the geometry of the monomers, thermal corrections of monomers and dimers, the contribution of spin–orbit coupling to monomers and dimers, and finally, the accuracy of the electronic energy correction calculated at different levels of theory. We find that optimizing the structures either at the ωB97X-D3BJ/aug-cc-pVTZ-PP or the M06-2X/aug-cc-pVTZ-PP level achieves the best thermal contribution to the binding free energy. The electronic energy correction can then be calculated at the ZORA-DLPNO–CCSD(T0) level with the SARC-ZORA-TZVPP basis for iodine and ma-ZORA-def2-TZVPP for non-iodine atoms.
We applied this methodology to calculate the binding free energies of iodine-containing dimer clusters, where we confirm the qualitative trends observed in previous studies. However, we identify that previous studies overestimate the stability of the clusters by several kcal/mol due to the neglect of relativistic effects. This means that their contributions to the currently studied nucleation pathways of new particle formation are likely overestimated.
Full citation:
Engsvang, M., Wu, H., and Elm, J., Iodine Clusters in the Atmosphere I: Computational Benchmark and Dimer Formation of Oxyacids and Oxides, ACS Omega, 9 (29), 31521-31532 (2024). https://doi.org/10.1021/acsomega.4c01235
Yosef Knattrup, Jakub Kubečka, Haide Wu, Frank Jensen, and Jonas Elm
Atmospheric molecular clusters, the onset of secondary aerosol formation, are a major part of the current uncertainty in modern climate models. Quantum chemical (QC) methods are usually employed in a funneling approach to identify the lowest free energy cluster structures. However, the funneling approach highly depends on the accuracy of low-cost methods to ensure that important low-lying minima are not missed.
Here we present a reparameterized GFN1-xTB model based on the clusteromics I–V datasets for studying atmospheric molecular clusters (AMC), denoted AMC-xTB. The AMC-xTB model reduces the mean of electronic binding energy errors from 7–11.8 kcal mol−1 to roughly 0 kcal mol−1 and the root mean square deviation from 7.6–12.3 kcal mol−1 to 0.81–1.45 kcal mol−1. In addition, the minimum structures obtained with AMC-xTB are closer to the ωB97X-D/6-31++G(d,p) level of theory compared to GFN1-xTB.
We employ the new parameterization in two new configurational sampling workflows that include an additional meta-dynamics sampling step using CREST with the AMC-xTB model. The first workflow, denoted the “independent workflow”, is a commonly used funneling approach with an additional CREST step, and the second, the “improvement workflow”, is where the best configuration currently known in the literature is improved with a CREST + AMC-xTB step. Testing the new workflow we find configurations lower in free energy for all the literature clusters with the largest improvement being up to 21 kcal mol−1.
Lastly, by employing the improvement workflow we massively screened 288 new multi-acid–multi-base clusters containing up to 8 different species. For these new multi-acid–multi-base cluster systems we observe that the improvement workflow finds configurations lower in free energy for 245 out of 288 (85.1%) cluster structures. Most of the improvements are within 2 kcal mol−1, but we see improvements up to 8.3 kcal mol−1. Hence, we can recommend this new workflow based on the AMC-xTB model for future studies on atmospheric molecular clusters.
Full citation:
Knattrup, Y., Kubečka, J., Wu, H., Jensen, F. and Elm, J., Reparameterization of GFN1-xTB for atmospheric molecular clusters: applications to multi-acid–multi-base systems. RSC Advances, 14, 20048-20055 (2024). https://doi.org/10.1039/D4RA03021D.
Astrid Nørskov Pedersen, Yosef Knattrup, and Jonas Elm
The role of organic compounds in atmospheric new particle formation is difficult to disentangle due to the myriad of potentially important oxygenated organic molecules (OOMs) present in the atmosphere. Using state-of-the-art quantum chemical methods, we here employ a novel approach, denoted the “cluster-of-functional-groups” approach, for studying the involvement of OOMs in atmospheric cluster formation. Instead of the usual “trial-and-error” approach of testing the ability of experimentally identified OOMs to form stable clusters with other nucleation precursors, we here study which, and how many, intermolecular interactions are required in a given OOM to form stable clusters. In this manner we can reverse engineer the elusive structure of OOM candidates that might be involved in organic enhanced atmospheric cluster formation.
We calculated the binding free energies of all combinations of donor and acceptor organic functional groups to investigate which functional groups most preferentially bind with each other and with other nucleation precursors such as sulfuric acid and bases (ammonia, methyl-, dimethyl- and trimethylamine). We find that multiple carboxyl groups lead to substantially more stable clusters compared to all other combinations of functional groups. Employing cluster dynamics simulations, we investigate how a hypothetically OOM composed of multiple carboxyl groups can stabilize sulfuric acid–base clusters and provide recommendations for potential atmospheric multi-carboxylic acid tracer compounds that should be explicitly studied in the future.
The presented “cluster-of-functional-groups” approach is generally applicable and can be employed in many other applications, such as ion-induced nucleation and potentially in elucidating the structural patterns in molecules that facilitate ice nucleation.
Full citation:
Pedersen, A. N., Knattrup, Y., and Elm, J., A cluster-of-functional-groups approach for studying organic enhanced atmospheric cluster formation. Aerosol Research, 2, 123–134 (2024). https://doi.org/10.5194/ar-2-123-2024.
Jan Thøgersen, Fani Madzharova, Tobias Weidner, and Frank Jensen
The deep ultraviolet photochemistry of aqueous pyruvate is believed to have been essential to the origin of life, and near ultraviolet excitation of pyruvate in aqueous aerosols is assumed to contribute significantly to the photochemistry of the Earth’s atmosphere. However, the primary photochemistry of aqueous pyruvate is unknown.
Here we study the susceptibility of aqueous pyruvate to photodissociation by deep ultraviolet and near ultraviolet irradiation with femtosecond spectroscopy supported by density functional theory calculations. The primary photo-dynamics of the aqueous pyruvate show that upon deep-UV excitation at 200 nm, about one in five excited pyruvate anions have dissociated by decarboxylation 100 ps after the excitation, while the rest of the pyruvate anions return to the ground state. Upon near-UV photoexcitation at a wavelength of 340 nm, the dissociation yield of aqueous pyruvate 200 ps after the excitation is insignificant and no products are observed.
The experimental results are explained by our calculations, which show that aqueous pyruvate anions excited at 200 nm have sufficient excess energy for decarboxylation, whereas excitation at 340 nm provides the aqueous pyruvate anions with insufficient energy to overcome the decarboxylation barrier.
Full citation:
Thøgersen, J., Madzharova, F., Weidner, T. and Jensen F., Aqueous pyruvate partly dissociates under deep ultraviolet irradiation but is resilient to near ultraviolet excitation. Nat Commun 15, 1978 (2024). https://doi.org/10.1038/s41467-024-46309-5
Morten Engsvang, Yosef Knattrup, Jakub Kubečka, and Jonas Elm
To understand Arctic amplification, it is necessary to understand both the direct and indirect aerosol effect. Especially the indirect aerosol effect is important, due to the low background level of cloud condensation nuclei in the Arctic. Previous studies have shown how iodine oxyacids can contribute to the formation of aerosols in marine and polar areas, and we speculate that chlorine oxyacids, if present, could also contribute to particle formation. Recent measurements have observed the presence of chloric (CA) and perchloric acid (PA) in significant concentrations in the Arctic.
Using quantum chemical methods, we have studied the (acid)0–2(base)0–2 clusters, where the acid denotes CA, PA, or sulfuric acid (SA) and the base denotes ammonia, methylamine, dimethylamine, or trimethylamine. This allowed us to simulate the cluster formation potential of the chemical species.
We found PA to have a high nucleation potential but, due to low concentrations, should only be present as a minor constituent of nucleating clusters. However, at low temperatures during high concentration events, it can become a substantial additional contribution to SA-driven nucleation. Therefore, further measurements and studies of larger multicomponent clusters should be pursued in order to constrain the potential contribution of PA to Arctic nucleation.
Full citation:
Engsvang, M., Knattrup, Y., Kubečka, J. and Elm J., Chlorine Oxyacids Potentially Contribute to Arctic Aerosol Formation. Environ. Sci. Technol. Lett. 11 (2), 101-105 (2024), https://doi.org/10.1021/acs.estlett.3c00902
Mads Greisen Højlund, Alberto Zoccante and Ove Christiansen
We derive equations of motion for bivariational wave functions with orthogonal adaptive basis sets and specialize the formalism to the coupled cluster Ansatz. The equations are related to the biorthogonal case in a transparent way, and similarities and differences are analyzed. We show that the amplitude equations are identical in the orthogonal and biorthogonal formalisms, while the linear equations that determine the basis set time evolution differ by symmetrization.
Applying the orthogonal framework to the nuclear dynamics problem, we introduce and implement the orthogonal time-dependent modal vibrational coupled cluster (oTDMVCC) method and benchmark it against exact reference results for four triatomic molecules as well as a reduced-dimensional (5D) trans-bithiophene model. We confirm numerically that the biorthogonal TDMVCC hierarchy converges to the exact solution, while oTDMVCC does not.
The differences between TDMVCC and oTDMVCC are found to be small for three of the five cases, but we also identify one case where the formal deficiency of the oTDMVCC approach results in clear and visible errors relative to the exact result. For the remaining example, oTDMVCC exhibits rather modest but visible errors.
Full citation: Højlund, M. G., Zoccante, A. and Christiansen, O., Time-dependent coupled cluster with orthogonal adaptive basis functions: General formalism and application to the vibrational problem. J. Chem. Phys. 160 (2): 024105 (2024). https://doi.org/10.1063/5.0186000
Nicolai Machholdt Høyer and Ove Christiansen
We present a new quasi-direct quantum molecular dynamics computational method which offers a compromise between quantum dynamics using a precomputed potential energy surface (PES) and fully direct quantum dynamics. This method is termed the time-dependent adaptive density-guided approach (TD-ADGA) and is a method for constructing a PES on the fly during a dynamics simulation. This is achieved by acquisition of new single-point (SP) calculations and refitting of the PES, depending on the need of the dynamics. The TD-ADGA is a further development of the adaptive density-guided approach (ADGA) for PES construction where the placement of SPs is guided by the density of the nuclear wave function.
In TD-ADGA, the ADGA framework has been integrated into the time propagation of the time-dependent nuclear wave function and we use the reduced one-mode density of this wave function to guide when and where new SPs are placed. The PES is thus extended or updated if the wave function moves into new areas or if a certain area becomes more important. Here, we derive equations for the reduced one-mode density for the time-dependent Hartree (TDH) method and for multiconfiguration time-dependent Hartree (MCTDH) methods, but the TD-ADGA can be used with any time-dependent wave function method as long as a density is available.
The TD-ADGA method has been investigated on molecular systems containing single- and double-minimum potentials and on single-mode and multi-mode systems. We explore different approaches to handle the fact that the TD-ADGA involves a PES that changes during the computation and show how results can be obtained that are in very good agreement with results obtained by using an accurate reference PES. Dynamics with TD-ADGA is essentially a black box procedure, where only the initialization of the system and how to compute SPs must be provided. The TD-ADGA thus makes it easier to carry out quantum molecular dynamics and the quasi-direct framework opens up the possibility to compute quantum dynamics accurately for larger molecular systems.
Full citation:
Høyer, N. M. and Christiansen, O., Quasi-direct Quantum Molecular Dynamics: The Time-Dependent Adaptive Density-Guided Approach for Potential Energy Surface Construction, J. Chem. Theory and Computation 20 (2), 558-579 (2024), https://doi.org/10.1021/acs.jctc.3c00962
Ditte Thomsen, Emil Mark Iversen, Jane Tygesen Skønager, Yuanyuan Luo, Linjie Li, Pontus Roldin, Michael Priestley, Henrik B. Pedersen, Mattias Hallquist, Mikael Ehn, Merete Bilde and Marianne Glasius
This study investigates the effects of temperature and relative humidity (RH) on the formation of secondary organic aerosol (SOA) from Δ3-carene, a prevalent monoterpene in boreal forests. Dark ozonolysis experiments of 10 ppb Δ3-carene were conducted in the Aarhus University Research on Aerosol (AURA) atmospheric simulation chamber at temperatures of 0, 10, and 20 °C. Under dry conditions (RH < 2%), the SOA formation in terms of both particle number and mass concentration shows minimal temperature dependence. This is in contrast to previous findings at higher initial concentrations and suggests an effect of VOC loading for Δ3-carene. Interestingly, the mass fraction of key oxidation products (cis-3-caric acid, cis-3-caronic acid) exhibit a temperature dependence suggesting continuous condensation at lower temperatures, while evaporation and further reactions over time become more favourable at higher temperatures.
The oxygen-to-carbon ratios in the particle phase and the occurrence of highly oxygenated organic molecules (HOM) in the gas phase show modest increases with higher temperatures. Predictions from the Aerosol Dynamics and Gas- and Particle-Phase Chemistry Kinetic Multilayer Model (ADCHAM) agrees with the experimental results regarding both physical particle properties and aerosol composition considering the experimental uncertainties. At high RH (∼80%, 10 °C), a considerable increase in the particle nucleation rate and particle number concentration is observed compared to experiments under dry conditions. This is likely due to enhanced particle nucleation resulting from more stable cluster formation of water and inorganics at increased RH. However, RH does not affect the particle mass concentration.
Full citation:
Thomsen, D., Iversen, E. M, Skønager, J. T., Luo, Y. Li, L., Roldin, P., Priestley, M., Pedersen, H. B., Hallquist, M., Ehn, M., Bilde, M. and Glasius, M., The effect of temperature and relative humidity on secondary organic aerosol formation from ozonolysis of Δ3-carene, Environ. Sci.: Atmos., 4, 88-103 (2024), https://doi.org/10.1039/D3EA00128H.
Sarah Suda Petters, Eva Rosendal Kjærgaard, Freja Hasager, Andreas Massling, Marianne Glasius and Merete Bilde
The role of airborne nanoparticles in atmospheric chemistry and public health is largely controlled by particle size, morphology, surface composition, and coating. Aerosol mass spectrometry provides real-time chemical characterization of submicron atmospheric particles, but analysis of nanoplastics in complex aerosol mixtures such as sea spray is severely limited by challenges associated with separation and ionization of the aerosol matrix. Here we characterize the internal and external mixing state of synthetic sea spray aerosols spiked with 150 nm nanoplastics.
Aerosols generated from pneumatic atomization and from a sea spray tank are compared. A humidified tandem differential mobility analyzer is used as a size and hygroscopicity filter, resulting in separation of nanoplastics from sea spray, and an inline high-resolution time-of-flight aerosol mass spectrometer is used to characterize particle composition and ionization efficiency. The separation technique amplified the detection limit of the airborne nanoplastics. A salt coating was found on the nanoplastics with coating thickness increasing exponentially with increasing bulk solution salinity, which was varied from 0 to 40 g kg−1. Relative ionization efficiencies of polystyrene and sea salt chloride were 0.19 and 0.36, respectively. The growth-factor derived hygroscopicity of sea salt was 1.4 at 75% relative humidity.
These results underscore the importance of separating airborne nanoplastics from sea salt aerosol for detailed online characterization by aerosol mass spectrometry and characterization of salt coatings as a function of water composition. The surface coating of nanoplastic aerosols by salts can profoundly impact their surface chemistry, water uptake, and humidified particle size distributions in the atmosphere.
Full citation:
Petters, S., Kjærgaard, E., Hasager, F., Massling, A., Glasius, M., Bilde, M., Morphology and hygroscopicity of nanoplastics in sea spray, Phys. Chem. Chem. Phys., 25 (47), 32430-32442 (2023). https://doi.org/10.1039/D3CP03793B
Frederik Bader, David Lauvergnat and Ove Christiansen
The efficiency of quantum chemical simulations of nuclear motion can in many cases greatly benefit from the application of curvilinear coordinate systems. This is rooted in the fact that a set of smartly selected curvilinear coordinates may represent the motion naturally well, thus decreasing the couplings between motions in these coordinates.
In this study, we assess the validity of different Taylor expansion-based approximations of kinetic energy operators in a (curvilinear) polyspherical parametrization. To this end, we investigate the accuracy as well as the numerical performance of the approximations in time-independent vibrational coupled cluster and full vibrational interaction calculations for several test cases ranging from tri- to penta-atomic molecules.
We find that several of the proposed schemes reproduce the vibrational ground state and excitation energies to a decent accuracy, justifying their application in future investigations. Furthermore, due to the restricted mode coupling and their inherent sum-of-products form, the new approximations open up the possibility of treating large molecular systems with efficient vibrational coupled cluster schemes in general coordinates.
Full citation:
Balder, F., Lauvergnat, D., Christiansen, O., Vibrationally correlated calculations in polyspherical coordinates: Taylor expansion-based kinetic energy operators, J. Chem. Phys. 159 (21), 214107 (2023). https://doi.org/10.1063/5.0171912
Andreas Buchgraitz Jensen, Mads Greisen Højlund, Alberto Zoccante, Niels Kristian Madsen and Ove Christiansen
The computation of the nuclear quantum dynamics of molecules is challenging, requiring both accuracy and efficiency to be applicable to systems of interest. Recently, theories have been developed for employing time-dependent basis functions (denoted modals) with vibrational coupled cluster theory (TDMVCC). The TDMVCC method was introduced along with a pilot implementation, which illustrated good accuracy in benchmark computations.
In this paper, we report an efficient implementation of TDMVCC, covering the case where the wave function and Hamiltonian contain up to two-mode couplings. After a careful regrouping of terms, the wave function can be propagated with a cubic computational scaling with respect to the number of degrees of freedom.
We discuss the use of a restricted set of active one-mode basis functions for each mode, as well as two interesting limits: (i) the use of a full active basis where the variational modal determination amounts essentially to the variational determination of a time-dependent reference state for the cluster expansion; and (ii) the use of a single function as an active basis for some degrees of freedom. The latter case defines a hybrid TDMVCC/TDH (time-dependent Hartree) approach that can obtain even lower computational scaling.
The resulting computational scaling for hybrid and full TDMVCC[2] is illustrated for polyaromatic hydrocarbons with up to 264 modes. Finally, computations on the internal vibrational redistribution of benzoic acid (39 modes) are used to show the faster convergence of TDMVCC/TDH hybrid computations towards TDMVCC compared to simple neglect of some degrees of freedom.
Full citation:
Jensen, A. B., Højlund, M. G., Zoccante, A., Madsen, N. K., Christiansen, O., Efficient time-dependent vibrational coupled cluster computations with time-dependent basis sets at the two-mode coupling level: Full and hybrid TDMVCC[2], J. Chem. Phys. 159 (20), 204106 (2023). https://doi.org/10.1063/5.0175506
Haide Wu, Morten Engsvang, Yosef Knattrup, Jakub Kubečka and Jonas Elm
The nucleation process leading to the formation of new atmospheric particles plays a crucial role in aerosol research. Quantum chemical (QC) calculations can be used to model the early stages of aerosol formation, where atmospheric vapor molecules interact and form stable molecular clusters. However, QC calculations heavily depend on the chosen computational method, and when dealing with large systems, striking a balance between accuracy and computational cost becomes essential.
We benchmarked the binding energies and structures and found the B97-3c method to be a good compromise between the accuracy and computational cost for studying large cluster systems. Further, we carefully assessed configurational sampling procedures for targeting large atmospheric molecular clusters containing up to 30 molecules (approximately 2 nm in diameter) and proposed a funneling approach with highly improved accuracy. We find that several parallel ABCluster explorations lead to better guesses for the cluster global energy minimum structures than one long exploration.
This methodology allows us to bridge computational studies of molecular clusters, which typically reach only around 1 nm, with experimental studies that often measure particles larger than 2 nm. By employing this workflow, we searched for low-energy configurations of large sulfuric acid–ammonia and sulfuric acid–dimethylamine clusters. We find that the binding free energies of clusters containing dimethylamine are unequivocally more stable than those of the ammonia-containing clusters. Our improved configurational sampling protocol can in the future be applied to study the growth and dynamics of large clusters of arbitrary compositions.
Full citation:
Wu, H., Engsvang, M., Knattrup, Y., Kubečka, J., Elm, J., Improved Configurational Sampling Protocol for Large Atmospheric Molecular Clusters, ACS Omega 8 (47), 45065–45077 (2023). https://doi.org/10.1021/acsomega.3c06794
Jakub Kubečka, Vitus Besel, Ivo Neefjes, Yosef Knattrup, Theo Kurtén, Hanna Vehkamäki and Jonas Elm
Computational modeling of atmospheric molecular clusters requires a comprehensive understanding of their complex configurational spaces, interaction patterns, stabilities against fragmentation, and even dynamic behaviors. To address these needs, we introduce the Jammy Key framework, a collection of automated scripts that facilitate and streamline molecular cluster modeling workflows. Jammy Key handles file manipulations between varieties of integrated third-party programs. The framework is divided into three main functionalities: (1) Jammy Key for configurational sampling (JKCS) to perform systematic configurational sampling of molecular clusters, (2) Jammy Key for quantum chemistry (JKQC) to analyze commonly used quantum chemistry output files and facilitate database construction, handling, and analysis, and (3) Jammy Key for machine learning (JKML) to manage machine learning methods in optimizing molecular cluster modeling.
This automation and machine learning utilization significantly reduces manual labor, greatly speeds up the search for molecular cluster configurations, and thus increases the number of systems that can be studied. Following the example of the Atmospheric Cluster Database (ACDB) of Elm (ACS Omega, 4, 10965–10984, 2019), the molecular clusters modeled in our group using the Jammy Key framework have been stored in an improved online GitHub repository named ACDB 2.0. In this work, we present the Jammy Key package alongside its assorted applications, which underline its versatility. Using several illustrative examples, we discuss how to choose appropriate combinations of methodologies for treating particular cluster types, including reactive, multicomponent, charged, or radical clusters, as well as clusters containing flexible or multiconformer monomers or heavy atoms. Finally, we present a detailed example of using the tools for atmospheric acid–base clusters.
Kubečka, J., Besel, V., Neefjes, I., Knattrup, Y., Kurtén, T., Vehkamäki, H., Elm, J., Computational Tools for Handling Molecular Clusters: Configurational Sampling, Storage, Analysis, and Machine Learning, ACS Omega 8 (47), 45115–45128, (2023). https://doi.org/10.1021/acsomega.3c07412
All publications from the PIs are found by clicking the relevant PI.