Publications, which are results from Center for Chemistry of Clouds.
Emil Mark Iversen, Merete Bilde, Henrik B.Pedersen
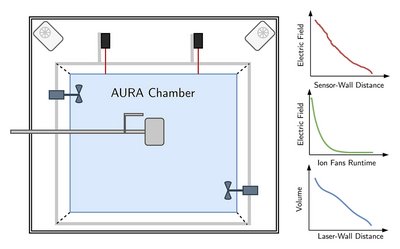
We describe three developments at the AURA atmospheric simulation chamber (made of Teflon and with a volume of ∼5 m3) aimed at an improved understanding of the physical conditions of the chamber to facilitate a better basis for comparisons between experimental data and results from numerical models. First, we demonstrate how the volume of the AURA chamber can be monitored by observing the position of a chamber wall using fixed laser distance sensors. The absolute volume calibration is obtained through a measurement of the relative humidity in the chamber during a controlled dilution experiment. Second, through a direct measurement, we characterize the occurrence, magnitude (∼0 – 80 kV/m), and decay time (∼10 – 20 h) of static electric fields inside the AURA chamber after charging. Further, we confirm directly that the AURA chamber can be significantly discharged and kept in a steady mode of charging with the addition of ion fans to the enclosure where the chamber is suspended. Third, we improve the air mixing capabilities at the AURA chamber by adding two mixing fans that allows efficient mixing of the chamber air within a few minutes. We characterize the effect of the electric field in the chamber and the rate of air mixing by direct measurements of the particle loss rate of injected polydisperse ammonium sulfate particles.
Full citation:
Iversen, M. E., Bilde, M., Pedersen, B. H., Developments at the AURA atmospheric simulation chamber to characterize chamber volume, air mixing, and charging,Aerosol Science and Technology, 1-23, (2024) https://doi.org/10.1080/02786826.2024.2429658
Lærke Sloth Nielsen, Tina Šantl-Temkiv, María Palomeque Sánchez, Andreas Massling, Josephine Caroline Ward, Pia Bomholt Jensen, Thomas Boesen, Markus Petters, Kai Finster, Merete Bilde, Bernadette Rosati
Airborne microorganisms impact cloud formation and are involved in disease spreading. The ability of airborne cells to survive and express genes may be limited by reduced water availability in the atmosphere and depend on the ability of the cells to attract water vapor at subsaturated conditions, i.e., their hygroscopicity. We assessed hygroscopic properties of the plant pathogen Pseudomonas syringae, known to participate in cloud formation. We used a hygroscopicity tandem differential mobility analyzer to examine both hydration and dehydration behavior in the relative humidity (RH) range 5–90%. The cells were aerosolized either from Milli-Q water or from a 35 g L–1 NaCl solution, resulting in pure cells or cells associated with NaCl. Pure cells exhibited no deliquescence/efflorescence and a small gradual water uptake reaching a maximum growth factor (GF) of 1.09 ± 0.01 at 90% RH. For cells associated with NaCl, we observed deliquescence and a much larger maximum GF of 1.74 ± 0.03 at 90% RH. Deliquescence RH was comparable to that of pure NaCl, highlighting the major role of the salt associated with the cells. It remains to be investigated how the observed hygroscopic properties relate to survival, metabolic, and ice-nucleation activities of airborne P. syringae.
Full citation:
Nielsen, L. S., Šantl-Temkiv, T., Sánchez, M. P., Massling, A., Ward, J. C., Jensen, P. B., Boesen, T., Petters, M., Finster, K., Bilde, M., Rosati, B., Water Uptake of Airborne Cells of P. syringae Measured with a Hygroscopicity Tandem Differential Mobility Analyzer, Environ. Sci. Technol., 58 (43), 19211–19221, (2024), https://doi.org/10.1021/acs.est.4c01817
Yuanyuan Luo, Ditte Thomsen, Emil Mark Iversen, Pontus Roldin, Jane Tygesen Skønager, Linjie Li, Michael Priestley, Henrik B. Pedersen, Mattias Hallquist, Merete Bilde, Marianne Glasius, and Mikael Ehn
Δ3-carene is a prominent monoterpene in the atmosphere, contributing significantly to secondary organic aerosol (SOA) formation. However, knowledge about Δ3-carene oxidation pathways, particularly regarding their ability to form highly oxygenated organic molecules (HOMs), is still limited. In this study, we present HOM measurements during Δ3-carene ozonolysis under various conditions in two simulation chambers. We identified numerous HOMs (monomers: C7−10H10−18O6−14; dimers: C17−20H24−34O6−18) using a chemical ionization mass spectrometer (CIMS). Δ3-carene ozonolysis yielded higher HOM concentrations than α-pinene, with a distinct distribution, indicating differences in formation pathways. All HOM signals decreased considerably at lower temperatures, reducing the estimated molar HOM yield from ∼ 3 % at 20 °C to ∼ 0.5 % at 0 °C. Interestingly, the temperature change altered the HOM distribution, increasing the observed dimer-to-monomer ratios from roughly 0.8 at 20 °C to 1.5 at 0 °C. HOM monomers with six or seven O atoms condensed more efficiently onto particles at colder temperatures, while monomers with nine or more O atoms and all dimers condensed irreversibly even at 20 °C. Using the gas- and particle-phase chemistry kinetic multilayer model ADCHAM, we were also able to reproduce the experimentally observed HOM composition, yields, and temperature dependence.
Full citation:
Luo, Y., Thomsen, D., Iversen, E. M., Roldin, P., Skønager, J. T., Li, L., Priestley, M., Pedersen, H. B., Hallquist, M., Bilde, M., Glasius, M., and Ehn, M.: Formation and temperature dependence of highly oxygenated organic molecules (HOMs) from Δ3-carene ozonolysis, Atmos. Chem. Phys., 24, 9459–9473 (2024). doi.org/10.5194/acp-24-9459-2024
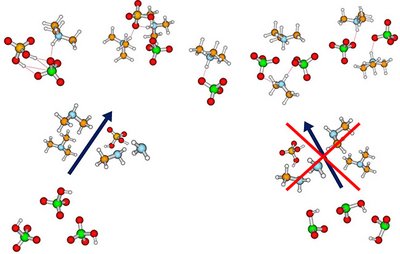
Andreas Buchgraitz Jensen and Jonas Elm
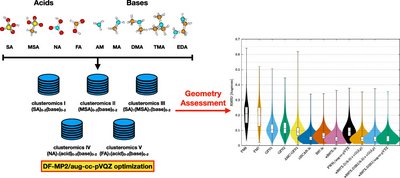
Atmospheric molecular clusters are important for the formation of new aerosol particles in the air. However, current experimental techniques are not able to yield direct insight into the cluster geometries. This implies that to date there is limited information about how accurately the applied computational methods depict the cluster structures. Here we massively benchmark the molecular geometries of atmospheric molecular clusters. We initially assessed how well different DF-MP2 approaches reproduce the geometries of 45 dimer clusters obtained at a high DF-CCSD(T)-F12b/cc-pVDZ-F12 level of theory. Based on the results, we find that the DF-MP2/aug-cc-pVQZ level of theory best resembles the DF-CCSD(T)-F12b/cc-pVDZ-F12 reference level. We subsequently optimized 1283 acid–base cluster structures (up to tetramers) at the DF-MP2/aug-cc-pVQZ level of theory and assessed how more approximate methods reproduce the geometries. Out of the tested semiempirical methods, we find that the newly parametrized atmospheric molecular cluster extended tight binding method (AMC-xTB) is most reliable for locating the correct lowest energy configuration and yields the lowest root mean square deviation (RMSD) compared to the reference level. In addition, we find that the DFT-3c methods show similar performance as the usually employed ωB97X-D/6-31++G(d,p) level of theory at a potentially reduced computational cost. This suggests that these methods could prove to be valuable for large-scale screening of cluster structures in the future.
Full citation:
Jensen, A. B., Elm, J., Massive Assessment of the Geometries of Atmospheric Molecular Clusters, J. Chem. Theory Comput., 20 (19), 8549–8558, (2024). https://doi.org/10.1021/acs.jctc.4c01046
Jakub Kubecka, Daniel Ayoubi, Zeyuan Tang, Yosef Knattrup, Morten Engsvang, Haide Wu and Jonas Elm
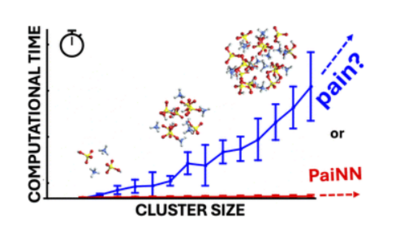
The computational cost of accurate quantum chemistry (QC) calculations of large molecular systems can often be unbearably high. Machine learning offers a lower computational cost compared to QC methods while maintaining their accuracy. In this study, we employ the polarizable atom interaction neural network (PaiNN) architecture to train and model the potential energy surface of molecular clusters relevant to atmospheric new particle formation, such as sulfuric acid–ammonia clusters. We compare the differences between PaiNN and previous kernel ridge regression modeling for the Clusteromics I–V data sets. We showcase three models capable of predicting electronic binding energies and interatomic forces with mean absolute errors of <0.3 kcal mol−1 and <0.2 kcal mol−1 Å−1 , respectively. Furthermore, we demonstrate that the error of the modeled properties remains below the chemical accuracy of 1 kcal mol−1 even for clusters vastly larger than those in the training database (up to (H2SO4)15(NH3)15 clusters, containing 30 molecules). Consequently, we emphasize the potential applications of these models for faster and more thorough configurational sampling and for boosting molecular dynamics studies of large atmospheric molecular clusters.
Full citation:
Kubecka, J., Ayoubi, D., Tang, Z., Knattrup, J., Engsvang, M., Wu, H., Elm, J., Accurate modeling of the potential energy surface of atmospheric molecular clusters boosted by neural networks, Environ. Sci.: Adv., 3 (10), 1438-1451 (2024), https://doi.org/10.1039/D4VA00255E
Eva R. Kjærgaard, Freja Hasager, Sarah S. Petters, Marianne Glasius, and Merete Bilde
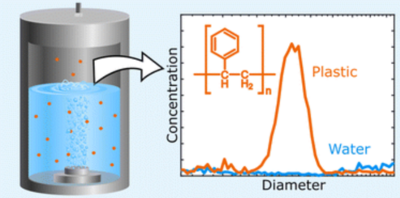
Micro- and nanoplastic particles have been detected in most environmental compartments. The presence of microplastics in the remote marine atmosphere and close to large lakes suggests bubble mediated water–air transfer as a source of airborne microplastics, however, quantitative estimates of plastic emission from surface waters remain uncertain. In this work, we elucidate the emission of submicron polystyrene nanospheres by bubble bursting in a laboratory setting from low salinity waters (salinity 0–1.0 g kg−1), polystyrene particle diameter (103, 147 and 269 nm), aqueous particle number concentrations in the range 4 × 107–2 × 109 cm−3, and bubble formation rate (0.88–3.35 L min−1 of air). Production of polystyrene aerosols was demonstrated using a scanning mobility particle sizer and confirmed by analysis of filter samples using pyrolysis gas chromatography coupled to mass spectrometry. We show that production of polystyrene aerosol particles scales linearly with the number concentration of plastic particles in the water. Our results suggest that small amounts (0.01 g kg−1) of salt increase polystyrene particle production. To the best of our knowledge this is the first study of bubble mediated water–air transfer of plastic particles as small as 100 nm.
Full citation:
Kjærgaard, E. R., Hasager, F., Petters, S. S., Glasius, M., Bilde, M., Bubble-mediated generation of airborne nanoplastic particles, Environ. Sci.: Processes Impacts, 26 (7), 1216-1226 (2024). https://doi.org/10.1039/D4EM00124A
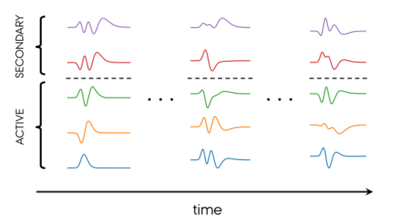
Mads Højlund and Ove Christiansen
We propose a new formulation of time-dependent coupled cluster with adaptive basis functions and division of the one-particle space into active and secondary subspaces. The formalism is fully bivariational in the sense of a real-valued time-dependent bivariational principle and converges to the complete-active-space solution, a property that is obtained by the use of biorthogonal basis functions. A key and distinguishing feature of the theory is that the active bra and ket functions span the same space by construction. This ensures numerical stability and is achieved by employing a split unitary/non-unitary basis set transformation: the unitary part changes the active space itself, while the non-unitary part transforms the active basis. The formulation covers vibrational as well as electron dynamics. Detailed equations of motion are derived and implemented in the context of vibrational dynamics, and the numerical behavior is studied and compared to related methods.
Højlund, M., Christiansen, O., A bivariational, stable, and convergent hierarchy for time-dependent coupled cluster with adaptive basis sets, J. Chem. Phys. 160 (17), 174119 (2024). https://doi.org/10.1063/5.0203914
Frederik Bader, David Lauvergnat, Ove Christiansen
Due to its efficiency and flexibility, the n-mode expansion is a frequently used tool for representing molecular potential energy surfaces in quantum chemical simulations. In this work, we investigate the performance of n-mode expansion-based models of kinetic energy operators in general polyspherical coordinate systems. In particular, we assess the operators with respect to accuracy in vibrationally correlated calculations and their effect on potential energy surface construction with the adaptive density guided approach. Our results show that the n-mode expansion-based operator variants are reliable and systematically improvable approximations of the full kinetic energy operator. Moreover, we introduce a workflow to generate the n-mode expanded kinetic energy operators on-the-fly within the adaptive density guided approach. This scheme can be applied in studies of species and coordinate systems, for which an analytical form of the kinetic energy operator is not available.
Full citation:
Bader, F., Lauvergnat, D., Christiansen, O., Efficient vibrationally correlated calculations using n-mode expansion-based kinetic energy operators, Phys. Chem. Chem. Phys., 26 (15), 11469-11481 (2024). https://doi.org/10.1039/D4CP00423J
Morten Engsvang, Haide Wu, and Jonas Elm
The contribution of iodine-containing compounds to atmospheric new particle formation is still not fully understood, but iodic acid and iodous acid are thought to be significant contributors. While several quantum chemical studies have been carried out on clusters containing iodine, there is no comprehensive benchmark study quantifying the accuracy of the applied methods. Here, we present the first study in a series that investigate the role of iodine species in atmospheric cluster formation. In this work, we have studied the iodic acid, iodous acid, iodine tetroxide, and iodine pentoxide monomers and their dimers formed with common atmospheric precursors.
We have tested the accuracy of commonly applied methods for calculating the geometry of the monomers, thermal corrections of monomers and dimers, the contribution of spin–orbit coupling to monomers and dimers, and finally, the accuracy of the electronic energy correction calculated at different levels of theory. We find that optimizing the structures either at the ωB97X-D3BJ/aug-cc-pVTZ-PP or the M06-2X/aug-cc-pVTZ-PP level achieves the best thermal contribution to the binding free energy. The electronic energy correction can then be calculated at the ZORA-DLPNO–CCSD(T0) level with the SARC-ZORA-TZVPP basis for iodine and ma-ZORA-def2-TZVPP for non-iodine atoms.
We applied this methodology to calculate the binding free energies of iodine-containing dimer clusters, where we confirm the qualitative trends observed in previous studies. However, we identify that previous studies overestimate the stability of the clusters by several kcal/mol due to the neglect of relativistic effects. This means that their contributions to the currently studied nucleation pathways of new particle formation are likely overestimated.
Full citation:
Engsvang, M., Wu, H., and Elm, J., Iodine Clusters in the Atmosphere I: Computational Benchmark and Dimer Formation of Oxyacids and Oxides, ACS Omega, 9 (29), 31521-31532 (2024). https://doi.org/10.1021/acsomega.4c01235
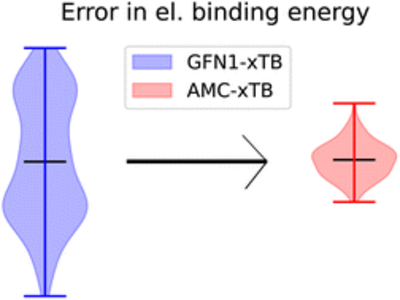
Yosef Knattrup, Jakub Kubečka, Haide Wu, Frank Jensen, and Jonas Elm
Atmospheric molecular clusters, the onset of secondary aerosol formation, are a major part of the current uncertainty in modern climate models. Quantum chemical (QC) methods are usually employed in a funneling approach to identify the lowest free energy cluster structures. However, the funneling approach highly depends on the accuracy of low-cost methods to ensure that important low-lying minima are not missed.
Here we present a reparameterized GFN1-xTB model based on the clusteromics I–V datasets for studying atmospheric molecular clusters (AMC), denoted AMC-xTB. The AMC-xTB model reduces the mean of electronic binding energy errors from 7–11.8 kcal mol−1 to roughly 0 kcal mol−1 and the root mean square deviation from 7.6–12.3 kcal mol−1 to 0.81–1.45 kcal mol−1. In addition, the minimum structures obtained with AMC-xTB are closer to the ωB97X-D/6-31++G(d,p) level of theory compared to GFN1-xTB.
We employ the new parameterization in two new configurational sampling workflows that include an additional meta-dynamics sampling step using CREST with the AMC-xTB model. The first workflow, denoted the “independent workflow”, is a commonly used funneling approach with an additional CREST step, and the second, the “improvement workflow”, is where the best configuration currently known in the literature is improved with a CREST + AMC-xTB step. Testing the new workflow we find configurations lower in free energy for all the literature clusters with the largest improvement being up to 21 kcal mol−1.
Lastly, by employing the improvement workflow we massively screened 288 new multi-acid–multi-base clusters containing up to 8 different species. For these new multi-acid–multi-base cluster systems we observe that the improvement workflow finds configurations lower in free energy for 245 out of 288 (85.1%) cluster structures. Most of the improvements are within 2 kcal mol−1, but we see improvements up to 8.3 kcal mol−1. Hence, we can recommend this new workflow based on the AMC-xTB model for future studies on atmospheric molecular clusters.
Full citation:
Knattrup, Y., Kubečka, J., Wu, H., Jensen, F. and Elm, J., Reparameterization of GFN1-xTB for atmospheric molecular clusters: applications to multi-acid–multi-base systems. RSC Advances, 14, 20048-20055 (2024). https://doi.org/10.1039/D4RA03021D.
Astrid Nørskov Pedersen, Yosef Knattrup, and Jonas Elm
The role of organic compounds in atmospheric new particle formation is difficult to disentangle due to the myriad of potentially important oxygenated organic molecules (OOMs) present in the atmosphere. Using state-of-the-art quantum chemical methods, we here employ a novel approach, denoted the “cluster-of-functional-groups” approach, for studying the involvement of OOMs in atmospheric cluster formation. Instead of the usual “trial-and-error” approach of testing the ability of experimentally identified OOMs to form stable clusters with other nucleation precursors, we here study which, and how many, intermolecular interactions are required in a given OOM to form stable clusters. In this manner we can reverse engineer the elusive structure of OOM candidates that might be involved in organic enhanced atmospheric cluster formation.
We calculated the binding free energies of all combinations of donor and acceptor organic functional groups to investigate which functional groups most preferentially bind with each other and with other nucleation precursors such as sulfuric acid and bases (ammonia, methyl-, dimethyl- and trimethylamine). We find that multiple carboxyl groups lead to substantially more stable clusters compared to all other combinations of functional groups. Employing cluster dynamics simulations, we investigate how a hypothetically OOM composed of multiple carboxyl groups can stabilize sulfuric acid–base clusters and provide recommendations for potential atmospheric multi-carboxylic acid tracer compounds that should be explicitly studied in the future.
The presented “cluster-of-functional-groups” approach is generally applicable and can be employed in many other applications, such as ion-induced nucleation and potentially in elucidating the structural patterns in molecules that facilitate ice nucleation.
Full citation:
Pedersen, A. N., Knattrup, Y., and Elm, J., A cluster-of-functional-groups approach for studying organic enhanced atmospheric cluster formation. Aerosol Research, 2, 123–134 (2024). https://doi.org/10.5194/ar-2-123-2024.
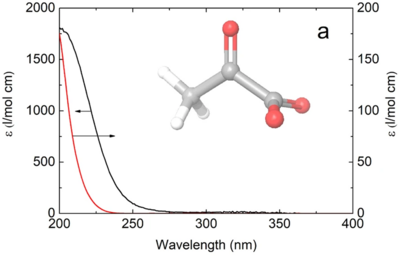
Jan Thøgersen, Fani Madzharova, Tobias Weidner, and Frank Jensen
The deep ultraviolet photochemistry of aqueous pyruvate is believed to have been essential to the origin of life, and near ultraviolet excitation of pyruvate in aqueous aerosols is assumed to contribute significantly to the photochemistry of the Earth’s atmosphere. However, the primary photochemistry of aqueous pyruvate is unknown.
Here we study the susceptibility of aqueous pyruvate to photodissociation by deep ultraviolet and near ultraviolet irradiation with femtosecond spectroscopy supported by density functional theory calculations. The primary photo-dynamics of the aqueous pyruvate show that upon deep-UV excitation at 200 nm, about one in five excited pyruvate anions have dissociated by decarboxylation 100 ps after the excitation, while the rest of the pyruvate anions return to the ground state. Upon near-UV photoexcitation at a wavelength of 340 nm, the dissociation yield of aqueous pyruvate 200 ps after the excitation is insignificant and no products are observed.
The experimental results are explained by our calculations, which show that aqueous pyruvate anions excited at 200 nm have sufficient excess energy for decarboxylation, whereas excitation at 340 nm provides the aqueous pyruvate anions with insufficient energy to overcome the decarboxylation barrier.
Full citation:
Thøgersen, J., Madzharova, F., Weidner, T. and Jensen F., Aqueous pyruvate partly dissociates under deep ultraviolet irradiation but is resilient to near ultraviolet excitation. Nat Commun 15, 1978 (2024). https://doi.org/10.1038/s41467-024-46309-5
Morten Engsvang, Yosef Knattrup, Jakub Kubečka, and Jonas Elm
To understand Arctic amplification, it is necessary to understand both the direct and indirect aerosol effect. Especially the indirect aerosol effect is important, due to the low background level of cloud condensation nuclei in the Arctic. Previous studies have shown how iodine oxyacids can contribute to the formation of aerosols in marine and polar areas, and we speculate that chlorine oxyacids, if present, could also contribute to particle formation. Recent measurements have observed the presence of chloric (CA) and perchloric acid (PA) in significant concentrations in the Arctic.
Using quantum chemical methods, we have studied the (acid)0–2(base)0–2 clusters, where the acid denotes CA, PA, or sulfuric acid (SA) and the base denotes ammonia, methylamine, dimethylamine, or trimethylamine. This allowed us to simulate the cluster formation potential of the chemical species.
We found PA to have a high nucleation potential but, due to low concentrations, should only be present as a minor constituent of nucleating clusters. However, at low temperatures during high concentration events, it can become a substantial additional contribution to SA-driven nucleation. Therefore, further measurements and studies of larger multicomponent clusters should be pursued in order to constrain the potential contribution of PA to Arctic nucleation.
Full citation:
Engsvang, M., Knattrup, Y., Kubečka, J. and Elm J., Chlorine Oxyacids Potentially Contribute to Arctic Aerosol Formation. Environ. Sci. Technol. Lett. 11 (2), 101-105 (2024), https://doi.org/10.1021/acs.estlett.3c00902
Mads Greisen Højlund, Alberto Zoccante and Ove Christiansen
We derive equations of motion for bivariational wave functions with orthogonal adaptive basis sets and specialize the formalism to the coupled cluster Ansatz. The equations are related to the biorthogonal case in a transparent way, and similarities and differences are analyzed. We show that the amplitude equations are identical in the orthogonal and biorthogonal formalisms, while the linear equations that determine the basis set time evolution differ by symmetrization.
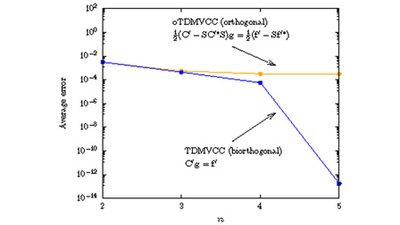
Applying the orthogonal framework to the nuclear dynamics problem, we introduce and implement the orthogonal time-dependent modal vibrational coupled cluster (oTDMVCC) method and benchmark it against exact reference results for four triatomic molecules as well as a reduced-dimensional (5D) trans-bithiophene model. We confirm numerically that the biorthogonal TDMVCC hierarchy converges to the exact solution, while oTDMVCC does not.
The differences between TDMVCC and oTDMVCC are found to be small for three of the five cases, but we also identify one case where the formal deficiency of the oTDMVCC approach results in clear and visible errors relative to the exact result. For the remaining example, oTDMVCC exhibits rather modest but visible errors.
Full citation: Højlund, M. G., Zoccante, A. and Christiansen, O., Time-dependent coupled cluster with orthogonal adaptive basis functions: General formalism and application to the vibrational problem. J. Chem. Phys. 160 (2): 024105 (2024). https://doi.org/10.1063/5.0186000
Nicolai Machholdt Høyer and Ove Christiansen
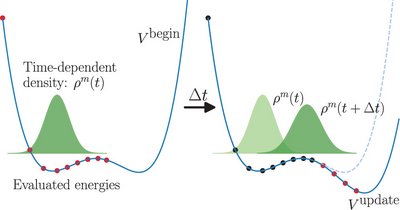
We present a new quasi-direct quantum molecular dynamics computational method which offers a compromise between quantum dynamics using a precomputed potential energy surface (PES) and fully direct quantum dynamics. This method is termed the time-dependent adaptive density-guided approach (TD-ADGA) and is a method for constructing a PES on the fly during a dynamics simulation. This is achieved by acquisition of new single-point (SP) calculations and refitting of the PES, depending on the need of the dynamics. The TD-ADGA is a further development of the adaptive density-guided approach (ADGA) for PES construction where the placement of SPs is guided by the density of the nuclear wave function.
In TD-ADGA, the ADGA framework has been integrated into the time propagation of the time-dependent nuclear wave function and we use the reduced one-mode density of this wave function to guide when and where new SPs are placed. The PES is thus extended or updated if the wave function moves into new areas or if a certain area becomes more important. Here, we derive equations for the reduced one-mode density for the time-dependent Hartree (TDH) method and for multiconfiguration time-dependent Hartree (MCTDH) methods, but the TD-ADGA can be used with any time-dependent wave function method as long as a density is available.
The TD-ADGA method has been investigated on molecular systems containing single- and double-minimum potentials and on single-mode and multi-mode systems. We explore different approaches to handle the fact that the TD-ADGA involves a PES that changes during the computation and show how results can be obtained that are in very good agreement with results obtained by using an accurate reference PES. Dynamics with TD-ADGA is essentially a black box procedure, where only the initialization of the system and how to compute SPs must be provided. The TD-ADGA thus makes it easier to carry out quantum molecular dynamics and the quasi-direct framework opens up the possibility to compute quantum dynamics accurately for larger molecular systems.
Full citation:
Høyer, N. M. and Christiansen, O., Quasi-direct Quantum Molecular Dynamics: The Time-Dependent Adaptive Density-Guided Approach for Potential Energy Surface Construction, J. Chem. Theory and Computation 20 (2), 558-579 (2024), https://doi.org/10.1021/acs.jctc.3c00962
Ditte Thomsen, Emil Mark Iversen, Jane Tygesen Skønager, Yuanyuan Luo, Linjie Li, Pontus Roldin, Michael Priestley, Henrik B. Pedersen, Mattias Hallquist, Mikael Ehn, Merete Bilde and Marianne Glasius
This study investigates the effects of temperature and relative humidity (RH) on the formation of secondary organic aerosol (SOA) from Δ3-carene, a prevalent monoterpene in boreal forests. Dark ozonolysis experiments of 10 ppb Δ3-carene were conducted in the Aarhus University Research on Aerosol (AURA) atmospheric simulation chamber at temperatures of 0, 10, and 20 °C. Under dry conditions (RH < 2%), the SOA formation in terms of both particle number and mass concentration shows minimal temperature dependence. This is in contrast to previous findings at higher initial concentrations and suggests an effect of VOC loading for Δ3-carene. Interestingly, the mass fraction of key oxidation products (cis-3-caric acid, cis-3-caronic acid) exhibit a temperature dependence suggesting continuous condensation at lower temperatures, while evaporation and further reactions over time become more favourable at higher temperatures.
The oxygen-to-carbon ratios in the particle phase and the occurrence of highly oxygenated organic molecules (HOM) in the gas phase show modest increases with higher temperatures. Predictions from the Aerosol Dynamics and Gas- and Particle-Phase Chemistry Kinetic Multilayer Model (ADCHAM) agrees with the experimental results regarding both physical particle properties and aerosol composition considering the experimental uncertainties. At high RH (∼80%, 10 °C), a considerable increase in the particle nucleation rate and particle number concentration is observed compared to experiments under dry conditions. This is likely due to enhanced particle nucleation resulting from more stable cluster formation of water and inorganics at increased RH. However, RH does not affect the particle mass concentration.
Full citation:
Thomsen, D., Iversen, E. M, Skønager, J. T., Luo, Y. Li, L., Roldin, P., Priestley, M., Pedersen, H. B., Hallquist, M., Ehn, M., Bilde, M. and Glasius, M., The effect of temperature and relative humidity on secondary organic aerosol formation from ozonolysis of Δ3-carene, Environ. Sci.: Atmos., 4, 88-103 (2024), https://doi.org/10.1039/D3EA00128H.
Sarah Suda Petters, Eva Rosendal Kjærgaard, Freja Hasager, Andreas Massling, Marianne Glasius and Merete Bilde
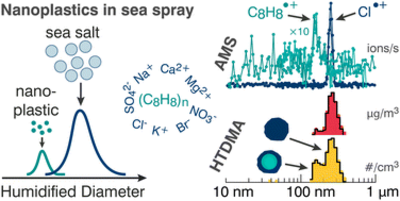
The role of airborne nanoparticles in atmospheric chemistry and public health is largely controlled by particle size, morphology, surface composition, and coating. Aerosol mass spectrometry provides real-time chemical characterization of submicron atmospheric particles, but analysis of nanoplastics in complex aerosol mixtures such as sea spray is severely limited by challenges associated with separation and ionization of the aerosol matrix. Here we characterize the internal and external mixing state of synthetic sea spray aerosols spiked with 150 nm nanoplastics.
Aerosols generated from pneumatic atomization and from a sea spray tank are compared. A humidified tandem differential mobility analyzer is used as a size and hygroscopicity filter, resulting in separation of nanoplastics from sea spray, and an inline high-resolution time-of-flight aerosol mass spectrometer is used to characterize particle composition and ionization efficiency. The separation technique amplified the detection limit of the airborne nanoplastics. A salt coating was found on the nanoplastics with coating thickness increasing exponentially with increasing bulk solution salinity, which was varied from 0 to 40 g kg−1. Relative ionization efficiencies of polystyrene and sea salt chloride were 0.19 and 0.36, respectively. The growth-factor derived hygroscopicity of sea salt was 1.4 at 75% relative humidity.
These results underscore the importance of separating airborne nanoplastics from sea salt aerosol for detailed online characterization by aerosol mass spectrometry and characterization of salt coatings as a function of water composition. The surface coating of nanoplastic aerosols by salts can profoundly impact their surface chemistry, water uptake, and humidified particle size distributions in the atmosphere.
Full citation:
Petters, S., Kjærgaard, E., Hasager, F., Massling, A., Glasius, M., Bilde, M., Morphology and hygroscopicity of nanoplastics in sea spray, Phys. Chem. Chem. Phys., 25 (47), 32430-32442 (2023). https://doi.org/10.1039/D3CP03793B
Frederik Bader, David Lauvergnat and Ove Christiansen
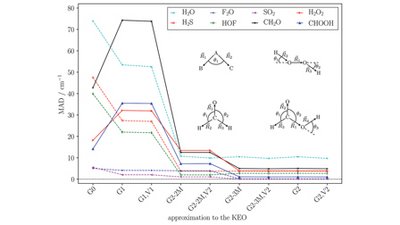
The efficiency of quantum chemical simulations of nuclear motion can in many cases greatly benefit from the application of curvilinear coordinate systems. This is rooted in the fact that a set of smartly selected curvilinear coordinates may represent the motion naturally well, thus decreasing the couplings between motions in these coordinates.
In this study, we assess the validity of different Taylor expansion-based approximations of kinetic energy operators in a (curvilinear) polyspherical parametrization. To this end, we investigate the accuracy as well as the numerical performance of the approximations in time-independent vibrational coupled cluster and full vibrational interaction calculations for several test cases ranging from tri- to penta-atomic molecules.
We find that several of the proposed schemes reproduce the vibrational ground state and excitation energies to a decent accuracy, justifying their application in future investigations. Furthermore, due to the restricted mode coupling and their inherent sum-of-products form, the new approximations open up the possibility of treating large molecular systems with efficient vibrational coupled cluster schemes in general coordinates.
Full citation:
Balder, F., Lauvergnat, D., Christiansen, O., Vibrationally correlated calculations in polyspherical coordinates: Taylor expansion-based kinetic energy operators, J. Chem. Phys. 159 (21), 214107 (2023). https://doi.org/10.1063/5.0171912
Andreas Buchgraitz Jensen, Mads Greisen Højlund, Alberto Zoccante, Niels Kristian Madsen and Ove Christiansen
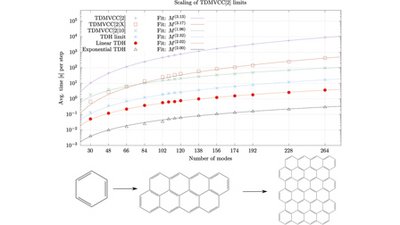
The computation of the nuclear quantum dynamics of molecules is challenging, requiring both accuracy and efficiency to be applicable to systems of interest. Recently, theories have been developed for employing time-dependent basis functions (denoted modals) with vibrational coupled cluster theory (TDMVCC). The TDMVCC method was introduced along with a pilot implementation, which illustrated good accuracy in benchmark computations.
In this paper, we report an efficient implementation of TDMVCC, covering the case where the wave function and Hamiltonian contain up to two-mode couplings. After a careful regrouping of terms, the wave function can be propagated with a cubic computational scaling with respect to the number of degrees of freedom.
We discuss the use of a restricted set of active one-mode basis functions for each mode, as well as two interesting limits: (i) the use of a full active basis where the variational modal determination amounts essentially to the variational determination of a time-dependent reference state for the cluster expansion; and (ii) the use of a single function as an active basis for some degrees of freedom. The latter case defines a hybrid TDMVCC/TDH (time-dependent Hartree) approach that can obtain even lower computational scaling.
The resulting computational scaling for hybrid and full TDMVCC[2] is illustrated for polyaromatic hydrocarbons with up to 264 modes. Finally, computations on the internal vibrational redistribution of benzoic acid (39 modes) are used to show the faster convergence of TDMVCC/TDH hybrid computations towards TDMVCC compared to simple neglect of some degrees of freedom.
Full citation:
Jensen, A. B., Højlund, M. G., Zoccante, A., Madsen, N. K., Christiansen, O., Efficient time-dependent vibrational coupled cluster computations with time-dependent basis sets at the two-mode coupling level: Full and hybrid TDMVCC[2], J. Chem. Phys. 159 (20), 204106 (2023). https://doi.org/10.1063/5.0175506
Haide Wu, Morten Engsvang, Yosef Knattrup, Jakub Kubečka and Jonas Elm
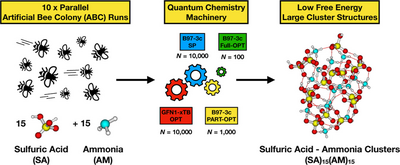
The nucleation process leading to the formation of new atmospheric particles plays a crucial role in aerosol research. Quantum chemical (QC) calculations can be used to model the early stages of aerosol formation, where atmospheric vapor molecules interact and form stable molecular clusters. However, QC calculations heavily depend on the chosen computational method, and when dealing with large systems, striking a balance between accuracy and computational cost becomes essential.
We benchmarked the binding energies and structures and found the B97-3c method to be a good compromise between the accuracy and computational cost for studying large cluster systems. Further, we carefully assessed configurational sampling procedures for targeting large atmospheric molecular clusters containing up to 30 molecules (approximately 2 nm in diameter) and proposed a funneling approach with highly improved accuracy. We find that several parallel ABCluster explorations lead to better guesses for the cluster global energy minimum structures than one long exploration.
This methodology allows us to bridge computational studies of molecular clusters, which typically reach only around 1 nm, with experimental studies that often measure particles larger than 2 nm. By employing this workflow, we searched for low-energy configurations of large sulfuric acid–ammonia and sulfuric acid–dimethylamine clusters. We find that the binding free energies of clusters containing dimethylamine are unequivocally more stable than those of the ammonia-containing clusters. Our improved configurational sampling protocol can in the future be applied to study the growth and dynamics of large clusters of arbitrary compositions.
Full citation:
Wu, H., Engsvang, M., Knattrup, Y., Kubečka, J., Elm, J., Improved Configurational Sampling Protocol for Large Atmospheric Molecular Clusters, ACS Omega 8 (47), 45065–45077 (2023). https://doi.org/10.1021/acsomega.3c06794
Jakub Kubečka, Vitus Besel, Ivo Neefjes, Yosef Knattrup, Theo Kurtén, Hanna Vehkamäki and Jonas Elm
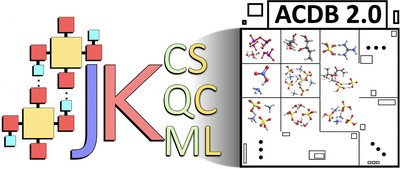
Computational modeling of atmospheric molecular clusters requires a comprehensive understanding of their complex configurational spaces, interaction patterns, stabilities against fragmentation, and even dynamic behaviors. To address these needs, we introduce the Jammy Key framework, a collection of automated scripts that facilitate and streamline molecular cluster modeling workflows. Jammy Key handles file manipulations between varieties of integrated third-party programs. The framework is divided into three main functionalities: (1) Jammy Key for configurational sampling (JKCS) to perform systematic configurational sampling of molecular clusters, (2) Jammy Key for quantum chemistry (JKQC) to analyze commonly used quantum chemistry output files and facilitate database construction, handling, and analysis, and (3) Jammy Key for machine learning (JKML) to manage machine learning methods in optimizing molecular cluster modeling.
This automation and machine learning utilization significantly reduces manual labor, greatly speeds up the search for molecular cluster configurations, and thus increases the number of systems that can be studied. Following the example of the Atmospheric Cluster Database (ACDB) of Elm (ACS Omega, 4, 10965–10984, 2019), the molecular clusters modeled in our group using the Jammy Key framework have been stored in an improved online GitHub repository named ACDB 2.0. In this work, we present the Jammy Key package alongside its assorted applications, which underline its versatility. Using several illustrative examples, we discuss how to choose appropriate combinations of methodologies for treating particular cluster types, including reactive, multicomponent, charged, or radical clusters, as well as clusters containing flexible or multiconformer monomers or heavy atoms. Finally, we present a detailed example of using the tools for atmospheric acid–base clusters.
Kubečka, J., Besel, V., Neefjes, I., Knattrup, Y., Kurtén, T., Vehkamäki, H., Elm, J., Computational Tools for Handling Molecular Clusters: Configurational Sampling, Storage, Analysis, and Machine Learning, ACS Omega 8 (47), 45115–45128, (2023). https://doi.org/10.1021/acsomega.3c07412
All publications from the PIs are found by clicking the relevant PI.